Jump to
S1C2
S1C3
Energy calculated at B1B95/6-31G*
| | hartrees |
|---|
| Energy at 0K | -1592.834163 |
| Energy at 298.15K | |
| HF Energy | -1592.834163 |
| Nuclear repulsion energy | 350.213979 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
518 |
492 |
0.00 |
51.11 |
0.07 |
0.12 |
| 2 |
A1 |
204 |
194 |
0.00 |
2.84 |
0.75 |
0.86 |
| 3 |
B1 |
452 |
429 |
0.00 |
34.98 |
0.75 |
0.86 |
| 4 |
B2 |
310 |
294 |
0.45 |
11.21 |
0.75 |
0.86 |
| 5 |
E |
436 |
414 |
0.09 |
7.43 |
0.75 |
0.86 |
| 5 |
E |
436 |
414 |
0.09 |
7.43 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1177.5 cm
-1
Scaled (by 0.9493) Zero Point Vibrational Energy (zpe) 1117.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/6-31G*
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.429 |
0.321 |
| S2 |
0.000 |
-1.429 |
0.321 |
| S3 |
-1.429 |
0.000 |
-0.321 |
| S4 |
1.429 |
0.000 |
-0.321 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.8589 | 2.1213 | 2.1213 |
S2 | 2.8589 | | 2.1213 | 2.1213 | S3 | 2.1213 | 2.1213 | | 2.8589 | S4 | 2.1213 | 2.1213 | 2.8589 | |
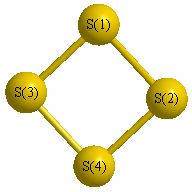 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
47.634 |
|
S1 |
S3 |
S4 |
47.634 |
| S2 |
S1 |
S3 |
47.634 |
|
S2 |
S4 |
S3 |
47.634 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.000 |
|
|
|
| 2 |
S |
0.000 |
|
|
|
| 3 |
S |
0.000 |
|
|
|
| 4 |
S |
0.000 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-49.594 |
0.000 |
0.000 |
| y |
0.000 |
-49.594 |
0.000 |
| z |
0.000 |
0.000 |
-54.696 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.551 |
0.000 |
0.000 |
| y |
0.000 |
2.551 |
0.000 |
| z |
0.000 |
0.000 |
-5.102 |
|
| Polar |
|---|
| 3z2-r2 | -10.203 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.260 |
0.000 |
0.000 |
| y |
0.000 |
10.260 |
0.000 |
| z |
0.000 |
0.000 |
5.369 |
<r2> (average value of r
2) Å
2
| <r2> |
169.425 |
| (<r2>)1/2 |
13.016 |
Jump to
S1C1
S1C3
Energy calculated at B1B95/6-31G*
| | hartrees |
|---|
| Energy at 0K | -1592.862681 |
| Energy at 298.15K | |
| HF Energy | -1592.862681 |
| Nuclear repulsion energy | 331.301460 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
693 |
658 |
2.79 |
30.45 |
0.23 |
0.37 |
| 2 |
A1 |
361 |
343 |
7.43 |
35.32 |
0.23 |
0.37 |
| 3 |
A1 |
109 |
103 |
0.90 |
10.36 |
0.49 |
0.66 |
| 4 |
A2 |
215 |
204 |
0.00 |
0.22 |
0.75 |
0.86 |
| 5 |
B2 |
668 |
634 |
135.55 |
2.78 |
0.75 |
0.86 |
| 6 |
B2 |
335 |
318 |
0.05 |
15.10 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1190.8 cm
-1
Scaled (by 0.9493) Zero Point Vibrational Energy (zpe) 1130.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.066 |
0.919 |
| S2 |
0.000 |
-1.066 |
0.919 |
| S3 |
0.000 |
1.597 |
-0.919 |
| S4 |
0.000 |
-1.597 |
-0.919 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.1325 | 1.9132 | 3.2357 |
S2 | 2.1325 | | 3.2357 | 1.9132 | S3 | 1.9132 | 3.2357 | | 3.1931 | S4 | 3.2357 | 1.9132 | 3.1931 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
106.092 |
|
S1 |
S3 |
S4 |
73.908 |
| S2 |
S1 |
S3 |
106.092 |
|
S2 |
S4 |
S3 |
73.908 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.101 |
|
|
|
| 2 |
S |
0.101 |
|
|
|
| 3 |
S |
-0.101 |
|
|
|
| 4 |
S |
-0.101 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.553 |
1.553 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-51.261 |
0.000 |
0.000 |
| y |
0.000 |
-53.964 |
0.000 |
| z |
0.000 |
0.000 |
-49.676 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.559 |
0.000 |
0.000 |
| y |
0.000 |
-3.495 |
0.000 |
| z |
0.000 |
0.000 |
2.936 |
|
| Polar |
|---|
| 3z2-r2 | 5.872 |
|---|
| x2-y2 | 2.703 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.335 |
0.000 |
0.000 |
| y |
0.000 |
17.648 |
0.000 |
| z |
0.000 |
0.000 |
11.048 |
<r2> (average value of r
2) Å
2
| <r2> |
204.264 |
| (<r2>)1/2 |
14.292 |
Jump to
S1C1
S1C2
Energy calculated at B1B95/6-31G*
| | hartrees |
|---|
| Energy at 0K | -1592.859272 |
| Energy at 298.15K | |
| HF Energy | -1592.859272 |
| Nuclear repulsion energy | 334.170858 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
746 |
708 |
0.00 |
38.07 |
0.28 |
0.44 |
| 2 |
Ag |
290 |
276 |
0.00 |
34.38 |
0.29 |
0.44 |
| 3 |
Au |
248 |
236 |
0.00 |
0.00 |
0.00 |
0.00 |
| 4 |
B1u |
695 |
660 |
149.60 |
0.00 |
0.00 |
0.00 |
| 5 |
B2u |
100i |
95i |
19.43 |
0.00 |
0.00 |
0.00 |
| 6 |
B3g |
335 |
318 |
0.00 |
17.29 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1107.0 cm
-1
Scaled (by 0.9493) Zero Point Vibrational Energy (zpe) 1050.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/6-31G*
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.948 |
1.277 |
| S2 |
0.000 |
0.948 |
-1.277 |
| S3 |
0.000 |
-0.948 |
1.277 |
| S4 |
0.000 |
-0.948 |
-1.277 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.5540 | 1.8959 | 3.1808 |
S2 | 2.5540 | | 3.1808 | 1.8959 | S3 | 1.8959 | 3.1808 | | 2.5540 | S4 | 3.1808 | 1.8959 | 2.5540 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
90.000 |
|
S1 |
S3 |
S4 |
90.000 |
| S2 |
S1 |
S3 |
90.000 |
|
S2 |
S4 |
S3 |
90.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.000 |
|
|
|
| 2 |
S |
0.000 |
|
|
|
| 3 |
S |
0.000 |
|
|
|
| 4 |
S |
0.000 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-51.639 |
0.000 |
0.000 |
| y |
0.000 |
-49.052 |
0.000 |
| z |
0.000 |
0.000 |
-53.579 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.324 |
0.000 |
0.000 |
| y |
0.000 |
3.557 |
0.000 |
| z |
0.000 |
0.000 |
-3.234 |
|
| Polar |
|---|
| 3z2-r2 | -6.467 |
|---|
| x2-y2 | -2.587 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.295 |
0.000 |
0.000 |
| y |
0.000 |
11.616 |
0.000 |
| z |
0.000 |
0.000 |
16.738 |
<r2> (average value of r
2) Å
2
| <r2> |
193.998 |
| (<r2>)1/2 |
13.928 |