Jump to
S1C2
Vibrational Frequencies calculated at B1B95/CEP-31G
Geometric Data calculated at B1B95/CEP-31G
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B1B95/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -53.513804 |
| Energy at 298.15K | -53.522925 |
| Nuclear repulsion energy | 109.792084 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3196 |
3060 |
44.74 |
|
|
|
| 2 |
A1 |
3017 |
2889 |
94.97 |
|
|
|
| 3 |
A1 |
2928 |
2804 |
97.24 |
|
|
|
| 4 |
A1 |
1541 |
1476 |
0.04 |
|
|
|
| 5 |
A1 |
1502 |
1439 |
5.90 |
|
|
|
| 6 |
A1 |
1473 |
1411 |
0.92 |
|
|
|
| 7 |
A1 |
1241 |
1188 |
1.32 |
|
|
|
| 8 |
A1 |
1143 |
1095 |
0.59 |
|
|
|
| 9 |
A1 |
987 |
945 |
48.76 |
|
|
|
| 10 |
A1 |
452 |
433 |
1.20 |
|
|
|
| 11 |
A1 |
198 |
190 |
3.45 |
|
|
|
| 12 |
A2 |
3090 |
2959 |
0.00 |
|
|
|
| 13 |
A2 |
1491 |
1427 |
0.00 |
|
|
|
| 14 |
A2 |
1222 |
1170 |
0.00 |
|
|
|
| 15 |
A2 |
1149 |
1100 |
0.00 |
|
|
|
| 16 |
A2 |
202 |
193 |
0.00 |
|
|
|
| 17 |
A2 |
45 |
43 |
0.00 |
|
|
|
| 18 |
B1 |
3091 |
2959 |
191.23 |
|
|
|
| 19 |
B1 |
2974 |
2847 |
151.95 |
|
|
|
| 20 |
B1 |
1491 |
1428 |
25.05 |
|
|
|
| 21 |
B1 |
1162 |
1113 |
8.99 |
|
|
|
| 22 |
B1 |
1118 |
1070 |
10.03 |
|
|
|
| 23 |
B1 |
216 |
207 |
10.10 |
|
|
|
| 24 |
B1 |
71 |
68 |
0.87 |
|
|
|
| 25 |
B2 |
3195 |
3059 |
28.59 |
|
|
|
| 26 |
B2 |
3015 |
2887 |
92.31 |
|
|
|
| 27 |
B2 |
1512 |
1448 |
1.46 |
|
|
|
| 28 |
B2 |
1481 |
1418 |
27.68 |
|
|
|
| 29 |
B2 |
1437 |
1376 |
57.65 |
|
|
|
| 30 |
B2 |
1197 |
1146 |
218.29 |
|
|
|
| 31 |
B2 |
1151 |
1102 |
130.89 |
|
|
|
| 32 |
B2 |
968 |
927 |
0.64 |
|
|
|
| 33 |
B2 |
385 |
369 |
5.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24670.3 cm
-1
Scaled (by 0.9575) Zero Point Vibrational Energy (zpe) 23621.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/CEP-31G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.327 |
| H2 |
-0.907 |
0.000 |
0.976 |
| H3 |
0.907 |
0.000 |
0.976 |
| O4 |
0.000 |
1.140 |
-0.534 |
| O5 |
0.000 |
-1.140 |
-0.534 |
| C6 |
0.000 |
2.387 |
0.199 |
| C7 |
0.000 |
-2.387 |
0.199 |
| H8 |
0.000 |
3.186 |
-0.551 |
| H9 |
0.000 |
-3.186 |
-0.551 |
| H10 |
-0.901 |
2.486 |
0.836 |
| H11 |
0.901 |
2.486 |
0.836 |
| H12 |
0.901 |
-2.486 |
0.836 |
| H13 |
-0.901 |
-2.486 |
0.836 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
O4 |
O5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.1158 | 1.1158 | 1.4285 | 1.4285 | 2.3901 | 2.3901 | 3.3047 | 3.3047 | 2.6925 | 2.6925 | 2.6925 | 2.6925 |
H2 | 1.1158 | | 1.8150 | 2.0984 | 2.0984 | 2.6689 | 2.6689 | 3.6476 | 3.6476 | 2.4896 | 3.0772 | 3.0772 | 2.4896 | H3 | 1.1158 | 1.8150 | | 2.0984 | 2.0984 | 2.6689 | 2.6689 | 3.6476 | 3.6476 | 3.0772 | 2.4896 | 2.4896 | 3.0772 | O4 | 1.4285 | 2.0984 | 2.0984 | | 2.2804 | 1.4460 | 3.6022 | 2.0460 | 4.3263 | 2.1207 | 2.1207 | 3.9792 | 3.9792 | O5 | 1.4285 | 2.0984 | 2.0984 | 2.2804 | | 3.6022 | 1.4460 | 4.3263 | 2.0460 | 3.9792 | 3.9792 | 2.1207 | 2.1207 | C6 | 2.3901 | 2.6689 | 2.6689 | 1.4460 | 3.6022 | | 4.7733 | 1.0961 | 5.6230 | 1.1076 | 1.1076 | 4.9956 | 4.9956 | C7 | 2.3901 | 2.6689 | 2.6689 | 3.6022 | 1.4460 | 4.7733 | | 5.6230 | 1.0961 | 4.9956 | 4.9956 | 1.1076 | 1.1076 | H8 | 3.3047 | 3.6476 | 3.6476 | 2.0460 | 4.3263 | 1.0961 | 5.6230 | | 6.3722 | 1.7956 | 1.7956 | 5.9078 | 5.9078 | H9 | 3.3047 | 3.6476 | 3.6476 | 4.3263 | 2.0460 | 5.6230 | 1.0961 | 6.3722 | | 5.9078 | 5.9078 | 1.7956 | 1.7956 | H10 | 2.6925 | 2.4896 | 3.0772 | 2.1207 | 3.9792 | 1.1076 | 4.9956 | 1.7956 | 5.9078 | | 1.8021 | 5.2879 | 4.9713 | H11 | 2.6925 | 3.0772 | 2.4896 | 2.1207 | 3.9792 | 1.1076 | 4.9956 | 1.7956 | 5.9078 | 1.8021 | | 4.9713 | 5.2879 | H12 | 2.6925 | 3.0772 | 2.4896 | 3.9792 | 2.1207 | 4.9956 | 1.1076 | 5.9078 | 1.7956 | 5.2879 | 4.9713 | | 1.8021 | H13 | 2.6925 | 2.4896 | 3.0772 | 3.9792 | 2.1207 | 4.9956 | 1.1076 | 5.9078 | 1.7956 | 4.9713 | 5.2879 | 1.8021 | |
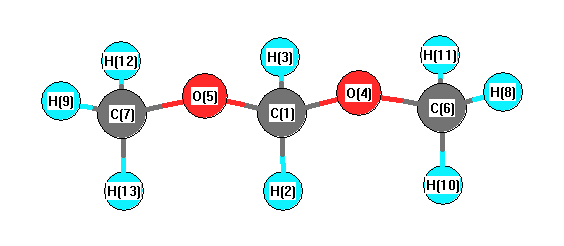 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C6 |
112.496 |
|
C1 |
O5 |
C7 |
112.496 |
| H2 |
C1 |
H3 |
108.840 |
|
H2 |
C1 |
O4 |
110.519 |
| H2 |
C1 |
O5 |
110.519 |
|
H3 |
C1 |
O4 |
110.519 |
| H3 |
C1 |
O5 |
110.519 |
|
O4 |
C1 |
O5 |
105.910 |
| O4 |
C6 |
H8 |
106.374 |
|
O4 |
C6 |
H10 |
111.613 |
| O4 |
C6 |
H11 |
111.613 |
|
O5 |
C7 |
H9 |
106.374 |
| O5 |
C7 |
H12 |
111.613 |
|
O5 |
C7 |
H13 |
111.613 |
| H8 |
C6 |
H10 |
109.142 |
|
H8 |
C6 |
H11 |
109.142 |
| H9 |
C7 |
H12 |
109.142 |
|
H9 |
C7 |
H13 |
109.142 |
| H10 |
C6 |
H11 |
108.886 |
|
H12 |
C7 |
H13 |
108.886 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/CEP-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.190 |
|
|
|
| 2 |
H |
0.137 |
|
|
|
| 3 |
H |
0.137 |
|
|
|
| 4 |
O |
-0.193 |
|
|
|
| 5 |
O |
-0.193 |
|
|
|
| 6 |
C |
-0.357 |
|
|
|
| 7 |
C |
-0.357 |
|
|
|
| 8 |
H |
0.220 |
|
|
|
| 9 |
H |
0.220 |
|
|
|
| 10 |
H |
0.144 |
|
|
|
| 11 |
H |
0.144 |
|
|
|
| 12 |
H |
0.144 |
|
|
|
| 13 |
H |
0.144 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.827 |
3.827 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.198 |
0.000 |
0.000 |
| y |
0.000 |
-22.701 |
0.000 |
| z |
0.000 |
0.000 |
-33.878 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.908 |
0.000 |
0.000 |
| y |
0.000 |
9.837 |
0.000 |
| z |
0.000 |
0.000 |
-6.929 |
|
| Polar |
|---|
| 3z2-r2 | -13.857 |
|---|
| x2-y2 | -8.497 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.765 |
0.000 |
0.000 |
| y |
0.000 |
7.246 |
0.000 |
| z |
0.000 |
0.000 |
4.784 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |