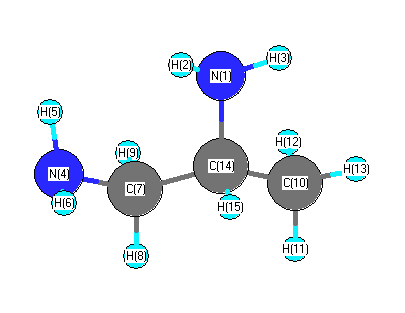
Vibrational Frequencies calculated at B1B95/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3732 |
3574 |
3.32 |
|
|
|
| 2 |
A |
3709 |
3551 |
6.36 |
|
|
|
| 3 |
A |
3598 |
3445 |
0.34 |
|
|
|
| 4 |
A |
3566 |
3414 |
11.61 |
|
|
|
| 5 |
A |
3165 |
3030 |
59.73 |
|
|
|
| 6 |
A |
3148 |
3014 |
63.97 |
|
|
|
| 7 |
A |
3131 |
2998 |
39.80 |
|
|
|
| 8 |
A |
3056 |
2926 |
40.43 |
|
|
|
| 9 |
A |
3001 |
2874 |
116.56 |
|
|
|
| 10 |
A |
2930 |
2805 |
134.07 |
|
|
|
| 11 |
A |
1677 |
1606 |
45.93 |
|
|
|
| 12 |
A |
1650 |
1580 |
67.81 |
|
|
|
| 13 |
A |
1506 |
1442 |
6.33 |
|
|
|
| 14 |
A |
1497 |
1434 |
10.73 |
|
|
|
| 15 |
A |
1491 |
1427 |
1.19 |
|
|
|
| 16 |
A |
1428 |
1367 |
1.54 |
|
|
|
| 17 |
A |
1413 |
1353 |
25.84 |
|
|
|
| 18 |
A |
1385 |
1327 |
23.36 |
|
|
|
| 19 |
A |
1367 |
1309 |
5.13 |
|
|
|
| 20 |
A |
1309 |
1253 |
8.45 |
|
|
|
| 21 |
A |
1261 |
1207 |
0.90 |
|
|
|
| 22 |
A |
1208 |
1157 |
13.98 |
|
|
|
| 23 |
A |
1179 |
1129 |
7.20 |
|
|
|
| 24 |
A |
1125 |
1077 |
27.21 |
|
|
|
| 25 |
A |
1047 |
1003 |
7.36 |
|
|
|
| 26 |
A |
1020 |
977 |
6.82 |
|
|
|
| 27 |
A |
964 |
923 |
2.85 |
|
|
|
| 28 |
A |
910 |
872 |
7.80 |
|
|
|
| 29 |
A |
850 |
814 |
7.10 |
|
|
|
| 30 |
A |
707 |
677 |
235.04 |
|
|
|
| 31 |
A |
625 |
598 |
291.96 |
|
|
|
| 32 |
A |
499 |
477 |
29.65 |
|
|
|
| 33 |
A |
460 |
440 |
5.89 |
|
|
|
| 34 |
A |
387 |
370 |
44.65 |
|
|
|
| 35 |
A |
329 |
315 |
29.53 |
|
|
|
| 36 |
A |
258 |
247 |
3.71 |
|
|
|
| 37 |
A |
229 |
219 |
11.72 |
|
|
|
| 38 |
A |
221 |
211 |
6.93 |
|
|
|
| 39 |
A |
146 |
140 |
2.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30590.3 cm
-1
Scaled (by 0.9575) Zero Point Vibrational Energy (zpe) 29290.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/CEP-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.570 |
|
|
|
| 2 |
H |
0.279 |
|
|
|
| 3 |
H |
0.240 |
|
|
|
| 4 |
N |
-0.527 |
|
|
|
| 5 |
H |
0.268 |
|
|
|
| 6 |
H |
0.250 |
|
|
|
| 7 |
C |
-0.331 |
|
|
|
| 8 |
H |
0.136 |
|
|
|
| 9 |
H |
0.136 |
|
|
|
| 10 |
C |
-0.447 |
|
|
|
| 11 |
H |
0.124 |
|
|
|
| 12 |
H |
0.145 |
|
|
|
| 13 |
H |
0.147 |
|
|
|
| 14 |
C |
0.036 |
|
|
|
| 15 |
H |
0.117 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.073 |
-0.896 |
1.282 |
1.897 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-28.360 |
1.956 |
1.111 |
| y |
1.956 |
-32.032 |
1.819 |
| z |
1.111 |
1.819 |
-32.634 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.972 |
1.956 |
1.111 |
| y |
1.956 |
-1.535 |
1.819 |
| z |
1.111 |
1.819 |
-2.438 |
|
| Polar |
|---|
| 3z2-r2 | -4.875 |
|---|
| x2-y2 | 3.671 |
|---|
| xy | 1.956 |
|---|
| xz | 1.111 |
|---|
| yz | 1.819 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.594 |
-0.139 |
0.149 |
| y |
-0.139 |
6.479 |
-0.125 |
| z |
0.149 |
-0.125 |
5.892 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |