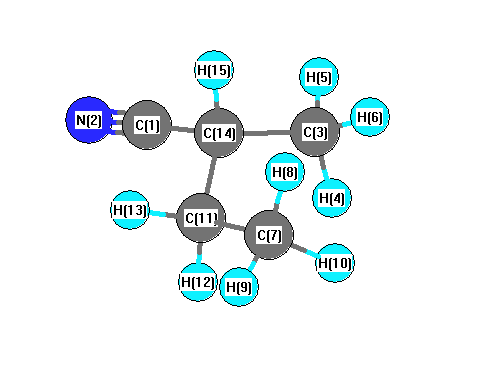
Vibrational Frequencies calculated at B1B95/CEP-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3179 |
3043 |
23.50 |
|
|
|
| 2 |
A |
3176 |
3040 |
28.01 |
|
|
|
| 3 |
A |
3166 |
3031 |
55.77 |
|
|
|
| 4 |
A |
3158 |
3023 |
41.20 |
|
|
|
| 5 |
A |
3137 |
3003 |
4.38 |
|
|
|
| 6 |
A |
3093 |
2961 |
19.57 |
|
|
|
| 7 |
A |
3088 |
2956 |
27.50 |
|
|
|
| 8 |
A |
3086 |
2954 |
0.24 |
|
|
|
| 9 |
A |
3076 |
2944 |
23.59 |
|
|
|
| 10 |
A |
2358 |
2257 |
21.30 |
|
|
|
| 11 |
A |
1500 |
1436 |
3.92 |
|
|
|
| 12 |
A |
1495 |
1431 |
11.14 |
|
|
|
| 13 |
A |
1493 |
1429 |
7.87 |
|
|
|
| 14 |
A |
1487 |
1423 |
14.38 |
|
|
|
| 15 |
A |
1478 |
1415 |
0.38 |
|
|
|
| 16 |
A |
1408 |
1348 |
3.52 |
|
|
|
| 17 |
A |
1403 |
1343 |
6.46 |
|
|
|
| 18 |
A |
1369 |
1311 |
0.98 |
|
|
|
| 19 |
A |
1332 |
1275 |
1.24 |
|
|
|
| 20 |
A |
1300 |
1245 |
1.54 |
|
|
|
| 21 |
A |
1262 |
1208 |
0.96 |
|
|
|
| 22 |
A |
1168 |
1118 |
2.42 |
|
|
|
| 23 |
A |
1146 |
1097 |
0.28 |
|
|
|
| 24 |
A |
1100 |
1053 |
6.15 |
|
|
|
| 25 |
A |
1056 |
1011 |
2.01 |
|
|
|
| 26 |
A |
996 |
953 |
2.49 |
|
|
|
| 27 |
A |
961 |
920 |
7.45 |
|
|
|
| 28 |
A |
902 |
863 |
0.67 |
|
|
|
| 29 |
A |
805 |
771 |
0.59 |
|
|
|
| 30 |
A |
753 |
721 |
5.85 |
|
|
|
| 31 |
A |
552 |
529 |
0.22 |
|
|
|
| 32 |
A |
527 |
504 |
1.26 |
|
|
|
| 33 |
A |
381 |
364 |
0.30 |
|
|
|
| 34 |
A |
310 |
297 |
0.32 |
|
|
|
| 35 |
A |
254 |
244 |
0.04 |
|
|
|
| 36 |
A |
211 |
202 |
0.08 |
|
|
|
| 37 |
A |
195 |
187 |
1.98 |
|
|
|
| 38 |
A |
156 |
149 |
4.74 |
|
|
|
| 39 |
A |
79 |
75 |
1.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28796.4 cm
-1
Scaled (by 0.9572) Zero Point Vibrational Energy (zpe) 27564.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/CEP-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.647 |
|
|
|
| 2 |
N |
0.383 |
|
|
|
| 3 |
C |
-0.528 |
|
|
|
| 4 |
H |
0.161 |
|
|
|
| 5 |
H |
0.184 |
|
|
|
| 6 |
H |
0.171 |
|
|
|
| 7 |
C |
-0.400 |
|
|
|
| 8 |
H |
0.132 |
|
|
|
| 9 |
H |
0.188 |
|
|
|
| 10 |
H |
0.134 |
|
|
|
| 11 |
C |
-0.303 |
|
|
|
| 12 |
H |
0.149 |
|
|
|
| 13 |
H |
0.164 |
|
|
|
| 14 |
C |
0.047 |
|
|
|
| 15 |
H |
0.164 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.975 |
1.282 |
0.547 |
4.212 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-48.267 |
-3.762 |
-0.892 |
| y |
-3.762 |
-36.665 |
-0.116 |
| z |
-0.892 |
-0.116 |
-35.840 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-12.015 |
-3.762 |
-0.892 |
| y |
-3.762 |
5.389 |
-0.116 |
| z |
-0.892 |
-0.116 |
6.626 |
|
| Polar |
|---|
| 3z2-r2 | 13.253 |
|---|
| x2-y2 | -11.603 |
|---|
| xy | -3.762 |
|---|
| xz | -0.892 |
|---|
| yz | -0.116 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.362 |
0.520 |
0.285 |
| y |
0.520 |
7.680 |
0.070 |
| z |
0.285 |
0.070 |
6.631 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |