Vibrational Frequencies calculated at B1B95/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3792 |
3634 |
75.46 |
|
|
|
| 2 |
A |
3593 |
3444 |
7.34 |
|
|
|
| 3 |
A |
3160 |
3029 |
16.32 |
|
|
|
| 4 |
A |
3134 |
3004 |
11.94 |
|
|
|
| 5 |
A |
3113 |
2983 |
23.73 |
|
|
|
| 6 |
A |
3107 |
2978 |
44.53 |
|
|
|
| 7 |
A |
3089 |
2961 |
17.75 |
|
|
|
| 8 |
A |
3057 |
2930 |
18.04 |
|
|
|
| 9 |
A |
2962 |
2839 |
71.54 |
|
|
|
| 10 |
A |
1840 |
1764 |
282.66 |
|
|
|
| 11 |
A |
1524 |
1460 |
0.34 |
|
|
|
| 12 |
A |
1506 |
1443 |
7.68 |
|
|
|
| 13 |
A |
1487 |
1426 |
0.70 |
|
|
|
| 14 |
A |
1457 |
1397 |
22.92 |
|
|
|
| 15 |
A |
1374 |
1317 |
15.79 |
|
|
|
| 16 |
A |
1368 |
1311 |
18.00 |
|
|
|
| 17 |
A |
1334 |
1278 |
4.80 |
|
|
|
| 18 |
A |
1326 |
1271 |
20.87 |
|
|
|
| 19 |
A |
1317 |
1263 |
3.95 |
|
|
|
| 20 |
A |
1256 |
1203 |
0.95 |
|
|
|
| 21 |
A |
1252 |
1200 |
3.38 |
|
|
|
| 22 |
A |
1223 |
1172 |
17.34 |
|
|
|
| 23 |
A |
1204 |
1154 |
6.79 |
|
|
|
| 24 |
A |
1169 |
1120 |
193.30 |
|
|
|
| 25 |
A |
1159 |
1110 |
55.40 |
|
|
|
| 26 |
A |
1108 |
1062 |
2.81 |
|
|
|
| 27 |
A |
1085 |
1040 |
4.06 |
|
|
|
| 28 |
A |
1010 |
968 |
4.37 |
|
|
|
| 29 |
A |
974 |
934 |
7.45 |
|
|
|
| 30 |
A |
943 |
904 |
5.92 |
|
|
|
| 31 |
A |
925 |
887 |
3.26 |
|
|
|
| 32 |
A |
901 |
863 |
65.29 |
|
|
|
| 33 |
A |
850 |
814 |
19.74 |
|
|
|
| 34 |
A |
781 |
748 |
2.12 |
|
|
|
| 35 |
A |
760 |
728 |
35.38 |
|
|
|
| 36 |
A |
675 |
647 |
88.44 |
|
|
|
| 37 |
A |
621 |
595 |
49.59 |
|
|
|
| 38 |
A |
594 |
569 |
12.80 |
|
|
|
| 39 |
A |
517 |
495 |
31.26 |
|
|
|
| 40 |
A |
498 |
477 |
13.83 |
|
|
|
| 41 |
A |
360 |
345 |
0.17 |
|
|
|
| 42 |
A |
247 |
237 |
3.69 |
|
|
|
| 43 |
A |
177 |
169 |
0.24 |
|
|
|
| 44 |
A |
59 |
57 |
0.11 |
|
|
|
| 45 |
A |
36 |
34 |
2.42 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31958.4 cm
-1
Scaled (by 0.9585) Zero Point Vibrational Energy (zpe) 30632.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
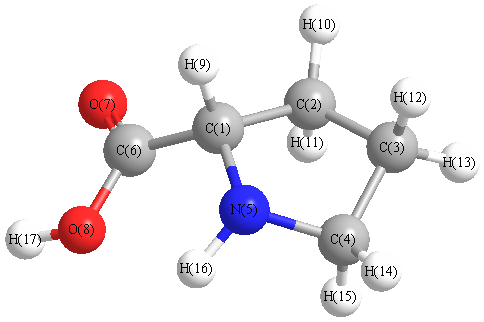
Charges from optimized geometry at B1B95/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.259 |
|
|
|
| 2 |
C |
-0.622 |
|
|
|
| 3 |
C |
-0.643 |
|
|
|
| 4 |
C |
-0.423 |
|
|
|
| 5 |
N |
-0.459 |
|
|
|
| 6 |
C |
0.663 |
|
|
|
| 7 |
O |
-0.796 |
|
|
|
| 8 |
O |
-0.375 |
|
|
|
| 9 |
H |
0.413 |
|
|
|
| 10 |
H |
0.262 |
|
|
|
| 11 |
H |
0.298 |
|
|
|
| 12 |
H |
0.195 |
|
|
|
| 13 |
H |
0.396 |
|
|
|
| 14 |
H |
0.394 |
|
|
|
| 15 |
H |
0.284 |
|
|
|
| 16 |
H |
-0.023 |
|
|
|
| 17 |
H |
0.176 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.237 |
-1.388 |
-0.352 |
1.452 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.909 |
5.980 |
-1.329 |
| y |
5.980 |
-49.489 |
2.555 |
| z |
-1.329 |
2.555 |
-47.536 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.604 |
5.980 |
-1.329 |
| y |
5.980 |
-2.267 |
2.555 |
| z |
-1.329 |
2.555 |
0.663 |
|
| Polar |
|---|
| 3z2-r2 | 1.326 |
|---|
| x2-y2 | 2.580 |
|---|
| xy | 5.980 |
|---|
| xz | -1.329 |
|---|
| yz | 2.555 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.660 |
-0.027 |
0.126 |
| y |
-0.027 |
11.013 |
0.123 |
| z |
0.126 |
0.123 |
9.505 |
<r2> (average value of r
2) Å
2
| <r2> |
253.568 |
| (<r2>)1/2 |
15.924 |