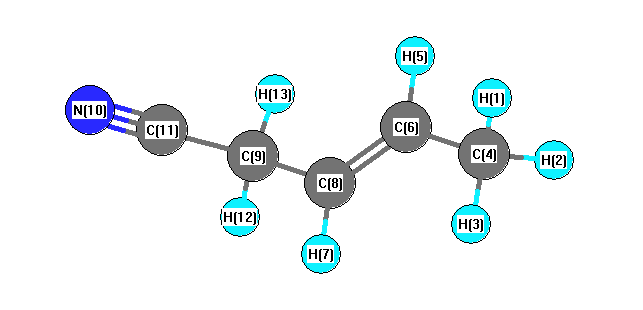
Vibrational Frequencies calculated at B1B95/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3179 |
3045 |
12.15 |
|
|
|
| 2 |
A |
3156 |
3023 |
26.20 |
|
|
|
| 3 |
A |
3140 |
3007 |
1.76 |
|
|
|
| 4 |
A |
3109 |
2978 |
12.96 |
|
|
|
| 5 |
A |
3106 |
2975 |
1.73 |
|
|
|
| 6 |
A |
3048 |
2919 |
6.20 |
|
|
|
| 7 |
A |
3046 |
2917 |
19.91 |
|
|
|
| 8 |
A |
2383 |
2282 |
10.82 |
|
|
|
| 9 |
A |
1765 |
1691 |
2.10 |
|
|
|
| 10 |
A |
1483 |
1420 |
9.78 |
|
|
|
| 11 |
A |
1472 |
1409 |
6.32 |
|
|
|
| 12 |
A |
1445 |
1384 |
8.40 |
|
|
|
| 13 |
A |
1404 |
1345 |
2.09 |
|
|
|
| 14 |
A |
1344 |
1287 |
1.95 |
|
|
|
| 15 |
A |
1327 |
1271 |
0.42 |
|
|
|
| 16 |
A |
1288 |
1234 |
7.07 |
|
|
|
| 17 |
A |
1215 |
1164 |
0.15 |
|
|
|
| 18 |
A |
1133 |
1085 |
0.45 |
|
|
|
| 19 |
A |
1091 |
1045 |
5.07 |
|
|
|
| 20 |
A |
1063 |
1018 |
0.91 |
|
|
|
| 21 |
A |
1011 |
968 |
25.06 |
|
|
|
| 22 |
A |
952 |
912 |
7.24 |
|
|
|
| 23 |
A |
944 |
904 |
6.40 |
|
|
|
| 24 |
A |
902 |
864 |
0.54 |
|
|
|
| 25 |
A |
772 |
739 |
0.69 |
|
|
|
| 26 |
A |
569 |
545 |
0.31 |
|
|
|
| 27 |
A |
452 |
432 |
0.66 |
|
|
|
| 28 |
A |
393 |
376 |
1.00 |
|
|
|
| 29 |
A |
293 |
281 |
1.39 |
|
|
|
| 30 |
A |
262 |
251 |
4.44 |
|
|
|
| 31 |
A |
211 |
202 |
1.01 |
|
|
|
| 32 |
A |
135 |
129 |
3.96 |
|
|
|
| 33 |
A |
72 |
69 |
2.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23581.6 cm
-1
Scaled (by 0.9577) Zero Point Vibrational Energy (zpe) 22584.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.142 |
|
|
|
| 2 |
H |
0.146 |
|
|
|
| 3 |
H |
0.138 |
|
|
|
| 4 |
C |
-0.447 |
|
|
|
| 5 |
H |
0.103 |
|
|
|
| 6 |
C |
-0.047 |
|
|
|
| 7 |
H |
0.119 |
|
|
|
| 8 |
C |
-0.050 |
|
|
|
| 9 |
C |
-0.394 |
|
|
|
| 10 |
N |
-0.322 |
|
|
|
| 11 |
C |
0.273 |
|
|
|
| 12 |
H |
0.166 |
|
|
|
| 13 |
H |
0.172 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.600 |
2.192 |
0.309 |
4.227 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.929 |
-6.054 |
-1.718 |
| y |
-6.054 |
-36.103 |
-1.148 |
| z |
-1.718 |
-1.148 |
-34.402 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-9.676 |
-6.054 |
-1.718 |
| y |
-6.054 |
3.562 |
-1.148 |
| z |
-1.718 |
-1.148 |
6.114 |
|
| Polar |
|---|
| 3z2-r2 | 12.228 |
|---|
| x2-y2 | -8.825 |
|---|
| xy | -6.054 |
|---|
| xz | -1.718 |
|---|
| yz | -1.148 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.955 |
0.372 |
-0.738 |
| y |
0.372 |
6.950 |
-0.343 |
| z |
-0.738 |
-0.343 |
6.704 |
<r2> (average value of r
2) Å
2
| <r2> |
233.225 |
| (<r2>)1/2 |
15.272 |