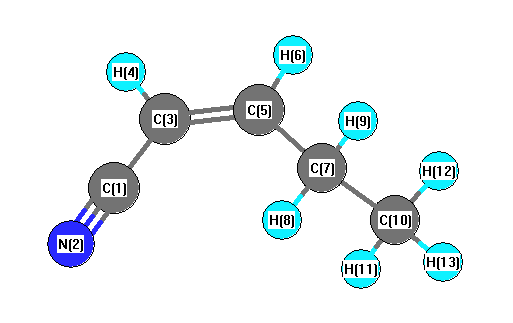
Vibrational Frequencies calculated at B1B95/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3209 |
3073 |
2.95 |
|
|
|
| 2 |
A |
3170 |
3036 |
9.03 |
|
|
|
| 3 |
A |
3152 |
3018 |
18.37 |
|
|
|
| 4 |
A |
3143 |
3010 |
20.21 |
|
|
|
| 5 |
A |
3109 |
2978 |
1.02 |
|
|
|
| 6 |
A |
3060 |
2930 |
25.06 |
|
|
|
| 7 |
A |
3043 |
2914 |
10.82 |
|
|
|
| 8 |
A |
2356 |
2257 |
16.29 |
|
|
|
| 9 |
A |
1708 |
1636 |
17.90 |
|
|
|
| 10 |
A |
1500 |
1437 |
5.89 |
|
|
|
| 11 |
A |
1493 |
1429 |
5.70 |
|
|
|
| 12 |
A |
1473 |
1410 |
4.77 |
|
|
|
| 13 |
A |
1416 |
1357 |
0.57 |
|
|
|
| 14 |
A |
1398 |
1339 |
0.66 |
|
|
|
| 15 |
A |
1333 |
1276 |
2.87 |
|
|
|
| 16 |
A |
1289 |
1234 |
0.01 |
|
|
|
| 17 |
A |
1246 |
1193 |
0.57 |
|
|
|
| 18 |
A |
1138 |
1090 |
0.55 |
|
|
|
| 19 |
A |
1087 |
1041 |
2.57 |
|
|
|
| 20 |
A |
1043 |
999 |
4.01 |
|
|
|
| 21 |
A |
1003 |
961 |
1.09 |
|
|
|
| 22 |
A |
968 |
927 |
4.44 |
|
|
|
| 23 |
A |
873 |
836 |
4.12 |
|
|
|
| 24 |
A |
802 |
768 |
6.58 |
|
|
|
| 25 |
A |
771 |
739 |
27.98 |
|
|
|
| 26 |
A |
673 |
645 |
2.26 |
|
|
|
| 27 |
A |
580 |
555 |
0.77 |
|
|
|
| 28 |
A |
405 |
387 |
0.38 |
|
|
|
| 29 |
A |
368 |
352 |
0.11 |
|
|
|
| 30 |
A |
243 |
233 |
0.70 |
|
|
|
| 31 |
A |
214 |
205 |
2.98 |
|
|
|
| 32 |
A |
147 |
141 |
4.72 |
|
|
|
| 33 |
A |
63 |
60 |
1.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23736.9 cm
-1
Scaled (by 0.9577) Zero Point Vibrational Energy (zpe) 22732.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.284 |
|
|
|
| 2 |
N |
-0.347 |
|
|
|
| 3 |
C |
-0.216 |
|
|
|
| 4 |
H |
0.149 |
|
|
|
| 5 |
C |
-0.015 |
|
|
|
| 6 |
H |
0.127 |
|
|
|
| 7 |
C |
-0.278 |
|
|
|
| 8 |
H |
0.149 |
|
|
|
| 9 |
H |
0.137 |
|
|
|
| 10 |
C |
-0.403 |
|
|
|
| 11 |
H |
0.144 |
|
|
|
| 12 |
H |
0.131 |
|
|
|
| 13 |
H |
0.138 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.325 |
2.466 |
0.052 |
4.140 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.769 |
-5.497 |
0.966 |
| y |
-5.497 |
-35.345 |
-0.646 |
| z |
0.966 |
-0.646 |
-36.197 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.999 |
-5.497 |
0.966 |
| y |
-5.497 |
3.638 |
-0.646 |
| z |
0.966 |
-0.646 |
2.360 |
|
| Polar |
|---|
| 3z2-r2 | 4.720 |
|---|
| x2-y2 | -6.425 |
|---|
| xy | -5.497 |
|---|
| xz | 0.966 |
|---|
| yz | -0.646 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.768 |
0.953 |
0.519 |
| y |
0.953 |
8.325 |
-0.270 |
| z |
0.519 |
-0.270 |
5.829 |
<r2> (average value of r
2) Å
2
| <r2> |
199.088 |
| (<r2>)1/2 |
14.110 |