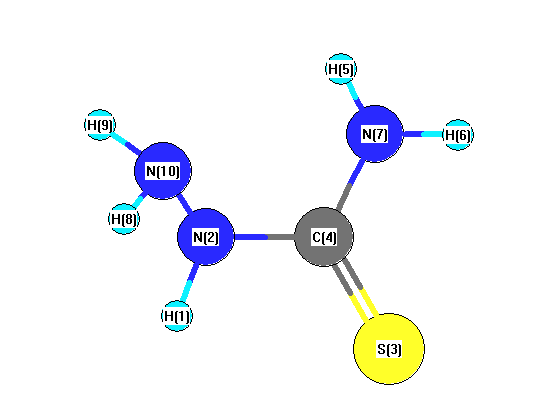
Vibrational Frequencies calculated at B1B95/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3786 |
3626 |
84.30 |
|
|
|
| 2 |
A |
3624 |
3471 |
25.00 |
|
|
|
| 3 |
A |
3613 |
3460 |
71.76 |
|
|
|
| 4 |
A |
3589 |
3437 |
6.24 |
|
|
|
| 5 |
A |
3503 |
3354 |
2.02 |
|
|
|
| 6 |
A |
1693 |
1622 |
46.44 |
|
|
|
| 7 |
A |
1610 |
1542 |
256.66 |
|
|
|
| 8 |
A |
1533 |
1468 |
191.71 |
|
|
|
| 9 |
A |
1465 |
1403 |
18.32 |
|
|
|
| 10 |
A |
1316 |
1260 |
0.43 |
|
|
|
| 11 |
A |
1297 |
1243 |
172.89 |
|
|
|
| 12 |
A |
1197 |
1146 |
23.72 |
|
|
|
| 13 |
A |
1034 |
990 |
31.43 |
|
|
|
| 14 |
A |
913 |
874 |
90.13 |
|
|
|
| 15 |
A |
836 |
800 |
46.06 |
|
|
|
| 16 |
A |
681 |
653 |
0.11 |
|
|
|
| 17 |
A |
618 |
592 |
0.70 |
|
|
|
| 18 |
A |
521 |
499 |
0.56 |
|
|
|
| 19 |
A |
505 |
483 |
25.05 |
|
|
|
| 20 |
A |
385 |
368 |
26.01 |
|
|
|
| 21 |
A |
381 |
364 |
183.46 |
|
|
|
| 22 |
A |
298 |
286 |
13.40 |
|
|
|
| 23 |
A |
180 |
173 |
48.73 |
|
|
|
| 24 |
A |
55 |
53 |
13.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17315.3 cm
-1
Scaled (by 0.9577) Zero Point Vibrational Energy (zpe) 16582.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.280 |
|
|
|
| 2 |
N |
-0.325 |
|
|
|
| 3 |
S |
-0.527 |
|
|
|
| 4 |
C |
0.474 |
|
|
|
| 5 |
H |
0.279 |
|
|
|
| 6 |
H |
0.293 |
|
|
|
| 7 |
N |
-0.543 |
|
|
|
| 8 |
H |
0.271 |
|
|
|
| 9 |
H |
0.271 |
|
|
|
| 10 |
N |
-0.474 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-5.900 |
0.534 |
0.035 |
5.924 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.997 |
4.968 |
-0.062 |
| y |
4.968 |
-30.352 |
-0.074 |
| z |
-0.062 |
-0.074 |
-37.848 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.897 |
4.968 |
-0.062 |
| y |
4.968 |
6.570 |
-0.074 |
| z |
-0.062 |
-0.074 |
-4.673 |
|
| Polar |
|---|
| 3z2-r2 | -9.347 |
|---|
| x2-y2 | -5.644 |
|---|
| xy | 4.968 |
|---|
| xz | -0.062 |
|---|
| yz | -0.074 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.232 |
-0.497 |
0.006 |
| y |
-0.497 |
7.533 |
-0.001 |
| z |
0.006 |
-0.001 |
4.731 |
<r2> (average value of r
2) Å
2
| <r2> |
159.431 |
| (<r2>)1/2 |
12.627 |