Vibrational Frequencies calculated at B1B95/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3509 |
3099 |
1.48 |
|
|
|
| 2 |
A' |
3497 |
3088 |
0.80 |
|
|
|
| 3 |
A' |
3369 |
2975 |
7.22 |
|
|
|
| 4 |
A' |
3361 |
2968 |
0.01 |
|
|
|
| 5 |
A' |
3337 |
2946 |
2.45 |
|
|
|
| 6 |
A' |
3324 |
2935 |
4.10 |
|
|
|
| 7 |
A' |
3305 |
2918 |
4.64 |
|
|
|
| 8 |
A' |
1698 |
1499 |
3.06 |
|
|
|
| 9 |
A' |
1690 |
1492 |
0.66 |
|
|
|
| 10 |
A' |
1688 |
1491 |
6.76 |
|
|
|
| 11 |
A' |
1676 |
1480 |
0.71 |
|
|
|
| 12 |
A' |
1666 |
1471 |
1.32 |
|
|
|
| 13 |
A' |
1577 |
1392 |
0.42 |
|
|
|
| 14 |
A' |
1549 |
1368 |
0.72 |
|
|
|
| 15 |
A' |
1542 |
1361 |
7.17 |
|
|
|
| 16 |
A' |
1466 |
1294 |
0.84 |
|
|
|
| 17 |
A' |
1365 |
1205 |
20.87 |
|
|
|
| 18 |
A' |
1220 |
1077 |
0.66 |
|
|
|
| 19 |
A' |
1167 |
1030 |
0.42 |
|
|
|
| 20 |
A' |
1131 |
999 |
1.48 |
|
|
|
| 21 |
A' |
1082 |
955 |
7.79 |
|
|
|
| 22 |
A' |
986 |
871 |
2.53 |
|
|
|
| 23 |
A' |
901 |
796 |
0.20 |
|
|
|
| 24 |
A' |
863 |
762 |
0.39 |
|
|
|
| 25 |
A' |
437 |
386 |
0.44 |
|
|
|
| 26 |
A' |
329 |
291 |
0.18 |
|
|
|
| 27 |
A' |
258 |
228 |
0.23 |
|
|
|
| 28 |
A' |
121 |
107 |
0.29 |
|
|
|
| 29 |
A" |
3511 |
3100 |
2.76 |
|
|
|
| 30 |
A" |
3480 |
3073 |
5.12 |
|
|
|
| 31 |
A" |
3465 |
3060 |
0.71 |
|
|
|
| 32 |
A" |
3464 |
3059 |
3.81 |
|
|
|
| 33 |
A" |
3431 |
3030 |
5.96 |
|
|
|
| 34 |
A" |
1685 |
1488 |
4.02 |
|
|
|
| 35 |
A" |
1671 |
1475 |
5.22 |
|
|
|
| 36 |
A" |
1436 |
1268 |
0.01 |
|
|
|
| 37 |
A" |
1411 |
1246 |
0.00 |
|
|
|
| 38 |
A" |
1343 |
1185 |
0.13 |
|
|
|
| 39 |
A" |
1176 |
1038 |
0.15 |
|
|
|
| 40 |
A" |
1070 |
945 |
1.42 |
|
|
|
| 41 |
A" |
1005 |
888 |
0.50 |
|
|
|
| 42 |
A" |
855 |
755 |
0.29 |
|
|
|
| 43 |
A" |
785 |
693 |
7.95 |
|
|
|
| 44 |
A" |
235 |
207 |
0.03 |
|
|
|
| 45 |
A" |
174 |
154 |
0.32 |
|
|
|
| 46 |
A" |
123 |
109 |
0.01 |
|
|
|
| 47 |
A" |
93 |
82 |
0.29 |
|
|
|
| 48 |
A" |
57 |
51 |
0.18 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 39290.9 cm
-1
Scaled (by 0.883) Zero Point Vibrational Energy (zpe) 34693.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
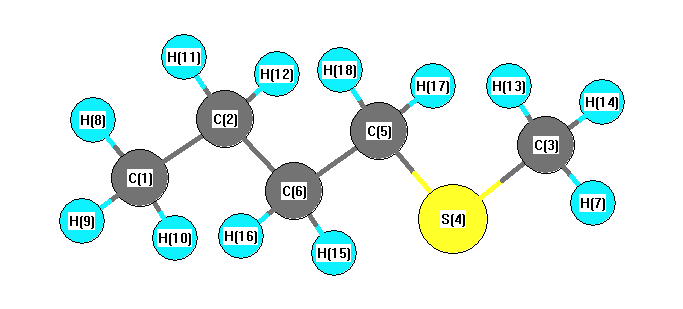
Charges from optimized geometry at B1B95/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.228 |
|
|
|
| 2 |
C |
-0.134 |
|
|
|
| 3 |
C |
-0.315 |
|
|
|
| 4 |
S |
0.138 |
|
|
|
| 5 |
C |
-0.219 |
|
|
|
| 6 |
C |
-0.138 |
|
|
|
| 7 |
H |
0.084 |
|
|
|
| 8 |
H |
0.076 |
|
|
|
| 9 |
H |
0.075 |
|
|
|
| 10 |
H |
0.075 |
|
|
|
| 11 |
H |
0.071 |
|
|
|
| 12 |
H |
0.071 |
|
|
|
| 13 |
H |
0.077 |
|
|
|
| 14 |
H |
0.077 |
|
|
|
| 15 |
H |
0.074 |
|
|
|
| 16 |
H |
0.074 |
|
|
|
| 17 |
H |
0.072 |
|
|
|
| 18 |
H |
0.072 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.641 |
0.614 |
0.000 |
0.888 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.101 |
1.209 |
0.000 |
| y |
1.209 |
-41.859 |
0.000 |
| z |
0.000 |
0.000 |
-43.352 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.496 |
1.209 |
0.000 |
| y |
1.209 |
1.868 |
0.000 |
| z |
0.000 |
0.000 |
-0.372 |
|
| Polar |
|---|
| 3z2-r2 | -0.745 |
|---|
| x2-y2 | -2.243 |
|---|
| xy | 1.209 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.274 |
1.192 |
0.000 |
| y |
1.192 |
5.658 |
0.000 |
| z |
0.000 |
0.000 |
3.911 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |