Vibrational Frequencies calculated at B1B95/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3122 |
2997 |
37.70 |
|
|
|
| 2 |
A' |
3055 |
2933 |
34.54 |
|
|
|
| 3 |
A' |
3043 |
2922 |
25.05 |
|
|
|
| 4 |
A' |
3028 |
2908 |
27.52 |
|
|
|
| 5 |
A' |
3019 |
2898 |
20.50 |
|
|
|
| 6 |
A' |
2880 |
2765 |
167.70 |
|
|
|
| 7 |
A' |
1842 |
1769 |
159.67 |
|
|
|
| 8 |
A' |
1504 |
1444 |
9.17 |
|
|
|
| 9 |
A' |
1489 |
1430 |
0.94 |
|
|
|
| 10 |
A' |
1480 |
1421 |
1.10 |
|
|
|
| 11 |
A' |
1442 |
1384 |
15.49 |
|
|
|
| 12 |
A' |
1412 |
1356 |
13.31 |
|
|
|
| 13 |
A' |
1405 |
1349 |
2.55 |
|
|
|
| 14 |
A' |
1399 |
1343 |
7.53 |
|
|
|
| 15 |
A' |
1358 |
1304 |
15.42 |
|
|
|
| 16 |
A' |
1265 |
1215 |
5.87 |
|
|
|
| 17 |
A' |
1138 |
1093 |
10.80 |
|
|
|
| 18 |
A' |
1084 |
1041 |
0.41 |
|
|
|
| 19 |
A' |
1048 |
1006 |
1.16 |
|
|
|
| 20 |
A' |
924 |
887 |
1.51 |
|
|
|
| 21 |
A' |
897 |
862 |
13.64 |
|
|
|
| 22 |
A' |
694 |
666 |
14.01 |
|
|
|
| 23 |
A' |
388 |
373 |
1.59 |
|
|
|
| 24 |
A' |
281 |
270 |
4.56 |
|
|
|
| 25 |
A' |
128 |
123 |
5.17 |
|
|
|
| 26 |
A" |
3116 |
2992 |
63.49 |
|
|
|
| 27 |
A" |
3097 |
2974 |
23.50 |
|
|
|
| 28 |
A" |
3059 |
2937 |
5.02 |
|
|
|
| 29 |
A" |
3046 |
2925 |
9.92 |
|
|
|
| 30 |
A" |
1492 |
1432 |
8.52 |
|
|
|
| 31 |
A" |
1321 |
1268 |
0.27 |
|
|
|
| 32 |
A" |
1301 |
1249 |
0.07 |
|
|
|
| 33 |
A" |
1227 |
1178 |
0.01 |
|
|
|
| 34 |
A" |
1153 |
1107 |
0.19 |
|
|
|
| 35 |
A" |
972 |
933 |
0.79 |
|
|
|
| 36 |
A" |
848 |
814 |
0.16 |
|
|
|
| 37 |
A" |
738 |
708 |
2.11 |
|
|
|
| 38 |
A" |
663 |
636 |
3.78 |
|
|
|
| 39 |
A" |
229 |
220 |
0.00 |
|
|
|
| 40 |
A" |
188 |
181 |
1.21 |
|
|
|
| 41 |
A" |
106 |
102 |
1.24 |
|
|
|
| 42 |
A" |
56 |
53 |
1.69 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30968.5 cm
-1
Scaled (by 0.9601) Zero Point Vibrational Energy (zpe) 29732.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
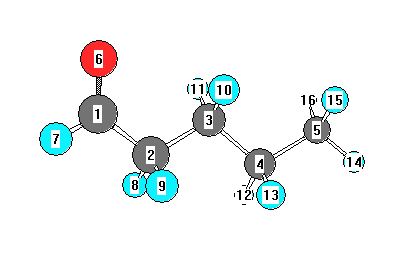
Charges from optimized geometry at B1B95/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.173 |
|
|
|
| 2 |
C |
-0.313 |
|
|
|
| 3 |
C |
-0.249 |
|
|
|
| 4 |
C |
-0.271 |
|
|
|
| 5 |
C |
-0.332 |
|
|
|
| 6 |
O |
-0.279 |
|
|
|
| 7 |
H |
0.084 |
|
|
|
| 8 |
H |
0.148 |
|
|
|
| 9 |
H |
0.148 |
|
|
|
| 10 |
H |
0.140 |
|
|
|
| 11 |
H |
0.140 |
|
|
|
| 12 |
H |
0.124 |
|
|
|
| 13 |
H |
0.124 |
|
|
|
| 14 |
H |
0.122 |
|
|
|
| 15 |
H |
0.121 |
|
|
|
| 16 |
H |
0.121 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.558 |
-0.123 |
0.000 |
2.561 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.704 |
3.863 |
0.000 |
| y |
3.863 |
-36.814 |
0.000 |
| z |
0.000 |
0.000 |
-37.249 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.672 |
3.863 |
0.000 |
| y |
3.863 |
4.163 |
0.000 |
| z |
0.000 |
0.000 |
3.510 |
|
| Polar |
|---|
| 3z2-r2 | 7.019 |
|---|
| x2-y2 | -7.890 |
|---|
| xy | 3.863 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.103 |
-0.414 |
0.000 |
| y |
-0.414 |
10.294 |
0.000 |
| z |
0.000 |
0.000 |
7.151 |
<r2> (average value of r
2) Å
2
| <r2> |
249.259 |
| (<r2>)1/2 |
15.788 |