Vibrational Frequencies calculated at B1B95/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3228 |
3082 |
0.00 |
|
|
|
| 2 |
Ag |
3201 |
3056 |
0.00 |
|
|
|
| 3 |
Ag |
3192 |
3048 |
0.00 |
|
|
|
| 4 |
Ag |
1629 |
1555 |
0.00 |
|
|
|
| 5 |
Ag |
1537 |
1468 |
0.00 |
|
|
|
| 6 |
Ag |
1471 |
1405 |
0.00 |
|
|
|
| 7 |
Ag |
1312 |
1253 |
0.00 |
|
|
|
| 8 |
Ag |
1190 |
1136 |
0.00 |
|
|
|
| 9 |
Ag |
1047 |
1000 |
0.00 |
|
|
|
| 10 |
Ag |
771 |
736 |
0.00 |
|
|
|
| 11 |
Ag |
626 |
598 |
0.00 |
|
|
|
| 12 |
Ag |
398 |
380 |
0.00 |
|
|
|
| 13 |
Au |
996 |
951 |
0.00 |
|
|
|
| 14 |
Au |
870 |
831 |
0.00 |
|
|
|
| 15 |
Au |
753 |
719 |
0.00 |
|
|
|
| 16 |
Au |
502 |
479 |
0.00 |
|
|
|
| 17 |
Au |
123 |
118 |
0.00 |
|
|
|
| 18 |
B1g |
967 |
923 |
0.00 |
|
|
|
| 19 |
B1g |
775 |
740 |
0.00 |
|
|
|
| 20 |
B1g |
484 |
463 |
0.00 |
|
|
|
| 21 |
B1g |
236 |
225 |
0.00 |
|
|
|
| 22 |
B1u |
3215 |
3070 |
55.43 |
|
|
|
| 23 |
B1u |
3196 |
3052 |
8.03 |
|
|
|
| 24 |
B1u |
3190 |
3046 |
17.63 |
|
|
|
| 25 |
B1u |
1705 |
1628 |
6.32 |
|
|
|
| 26 |
B1u |
1500 |
1432 |
1.26 |
|
|
|
| 27 |
B1u |
1348 |
1287 |
6.40 |
|
|
|
| 28 |
B1u |
1290 |
1231 |
7.23 |
|
|
|
| 29 |
B1u |
1176 |
1123 |
6.24 |
|
|
|
| 30 |
B1u |
908 |
867 |
1.54 |
|
|
|
| 31 |
B1u |
654 |
625 |
0.75 |
|
|
|
| 32 |
B1u |
226 |
216 |
1.22 |
|
|
|
| 33 |
B2g |
997 |
952 |
0.00 |
|
|
|
| 34 |
B2g |
914 |
872 |
0.00 |
|
|
|
| 35 |
B2g |
851 |
812 |
0.00 |
|
|
|
| 36 |
B2g |
775 |
740 |
0.00 |
|
|
|
| 37 |
B2g |
589 |
562 |
0.00 |
|
|
|
| 38 |
B2g |
271 |
258 |
0.00 |
|
|
|
| 39 |
B2u |
3228 |
3082 |
57.56 |
|
|
|
| 40 |
B2u |
3201 |
3056 |
0.16 |
|
|
|
| 41 |
B2u |
1612 |
1539 |
4.82 |
|
|
|
| 42 |
B2u |
1496 |
1429 |
1.68 |
|
|
|
| 43 |
B2u |
1453 |
1387 |
1.54 |
|
|
|
| 44 |
B2u |
1407 |
1344 |
3.39 |
|
|
|
| 45 |
B2u |
1199 |
1145 |
2.34 |
|
|
|
| 46 |
B2u |
1169 |
1117 |
1.32 |
|
|
|
| 47 |
B2u |
1043 |
996 |
4.73 |
|
|
|
| 48 |
B2u |
835 |
797 |
0.02 |
|
|
|
| 49 |
B2u |
607 |
579 |
8.37 |
|
|
|
| 50 |
B3g |
3215 |
3070 |
0.00 |
|
|
|
| 51 |
B3g |
3196 |
3052 |
0.00 |
|
|
|
| 52 |
B3g |
1706 |
1629 |
0.00 |
|
|
|
| 53 |
B3g |
1654 |
1579 |
0.00 |
|
|
|
| 54 |
B3g |
1417 |
1353 |
0.00 |
|
|
|
| 55 |
B3g |
1299 |
1240 |
0.00 |
|
|
|
| 56 |
B3g |
1213 |
1158 |
0.00 |
|
|
|
| 57 |
B3g |
1135 |
1084 |
0.00 |
|
|
|
| 58 |
B3g |
918 |
877 |
0.00 |
|
|
|
| 59 |
B3g |
523 |
499 |
0.00 |
|
|
|
| 60 |
B3g |
383 |
366 |
0.00 |
|
|
|
| 61 |
B3u |
972 |
928 |
5.72 |
|
|
|
| 62 |
B3u |
896 |
856 |
56.23 |
|
|
|
| 63 |
B3u |
744 |
711 |
64.26 |
|
|
|
| 64 |
B3u |
479 |
457 |
15.18 |
|
|
|
| 65 |
B3u |
385 |
368 |
0.08 |
|
|
|
| 66 |
B3u |
91 |
87 |
0.97 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42793.5 cm
-1
Scaled (by 0.9548) Zero Point Vibrational Energy (zpe) 40859.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
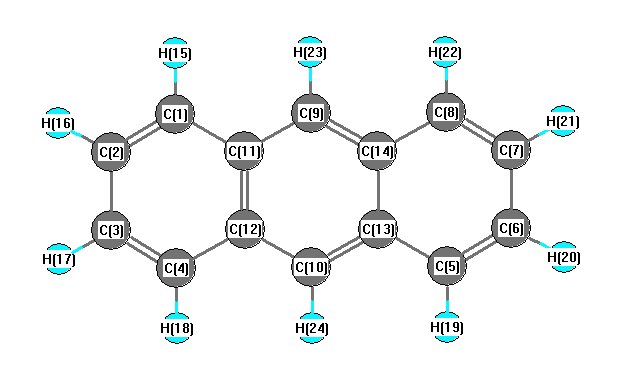
Charges from optimized geometry at B1B95/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.124 |
|
|
|
| 2 |
C |
-0.124 |
|
|
|
| 3 |
C |
-0.124 |
|
|
|
| 4 |
C |
-0.124 |
|
|
|
| 5 |
C |
-0.124 |
|
|
|
| 6 |
C |
-0.124 |
|
|
|
| 7 |
C |
-0.124 |
|
|
|
| 8 |
C |
-0.124 |
|
|
|
| 9 |
C |
-0.189 |
|
|
|
| 10 |
C |
-0.189 |
|
|
|
| 11 |
C |
0.066 |
|
|
|
| 12 |
C |
0.066 |
|
|
|
| 13 |
C |
0.066 |
|
|
|
| 14 |
C |
0.066 |
|
|
|
| 15 |
H |
0.110 |
|
|
|
| 16 |
H |
0.112 |
|
|
|
| 17 |
H |
0.112 |
|
|
|
| 18 |
H |
0.110 |
|
|
|
| 19 |
H |
0.110 |
|
|
|
| 20 |
H |
0.112 |
|
|
|
| 21 |
H |
0.112 |
|
|
|
| 22 |
H |
0.110 |
|
|
|
| 23 |
H |
0.108 |
|
|
|
| 24 |
H |
0.108 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-85.713 |
0.000 |
0.000 |
| y |
0.000 |
-68.719 |
0.000 |
| z |
0.000 |
0.000 |
-69.611 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-16.548 |
0.000 |
0.000 |
| y |
0.000 |
8.943 |
0.000 |
| z |
0.000 |
0.000 |
7.606 |
|
| Polar |
|---|
| 3z2-r2 | 15.211 |
|---|
| x2-y2 | -16.994 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.696 |
0.000 |
0.000 |
| y |
0.000 |
39.416 |
0.000 |
| z |
0.000 |
0.000 |
22.246 |
<r2> (average value of r
2) Å
2
| <r2> |
794.428 |
| (<r2>)1/2 |
28.186 |