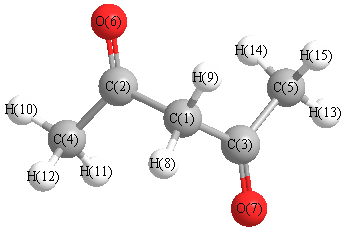
Vibrational Frequencies calculated at B1B95/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3209 |
3064 |
6.06 |
|
|
|
| 2 |
A |
3151 |
3008 |
4.29 |
|
|
|
| 3 |
A |
3120 |
2979 |
1.91 |
|
|
|
| 4 |
A |
3080 |
2941 |
0.33 |
|
|
|
| 5 |
A |
1866 |
1782 |
20.93 |
|
|
|
| 6 |
A |
1500 |
1432 |
7.90 |
|
|
|
| 7 |
A |
1497 |
1429 |
16.84 |
|
|
|
| 8 |
A |
1484 |
1417 |
8.31 |
|
|
|
| 9 |
A |
1410 |
1346 |
8.79 |
|
|
|
| 10 |
A |
1292 |
1234 |
26.93 |
|
|
|
| 11 |
A |
1155 |
1103 |
1.67 |
|
|
|
| 12 |
A |
1085 |
1036 |
0.87 |
|
|
|
| 13 |
A |
951 |
908 |
0.51 |
|
|
|
| 14 |
A |
803 |
767 |
0.19 |
|
|
|
| 15 |
A |
626 |
598 |
1.39 |
|
|
|
| 16 |
A |
488 |
466 |
6.73 |
|
|
|
| 17 |
A |
316 |
301 |
0.75 |
|
|
|
| 18 |
A |
174 |
166 |
0.19 |
|
|
|
| 19 |
A |
134 |
128 |
0.99 |
|
|
|
| 20 |
A |
61 |
58 |
7.22 |
|
|
|
| 21 |
B |
3209 |
3064 |
6.23 |
|
|
|
| 22 |
B |
3193 |
3049 |
5.17 |
|
|
|
| 23 |
B |
3150 |
3008 |
0.48 |
|
|
|
| 24 |
B |
3080 |
2940 |
2.20 |
|
|
|
| 25 |
B |
1840 |
1757 |
312.23 |
|
|
|
| 26 |
B |
1497 |
1429 |
1.40 |
|
|
|
| 27 |
B |
1493 |
1426 |
24.72 |
|
|
|
| 28 |
B |
1409 |
1346 |
105.40 |
|
|
|
| 29 |
B |
1268 |
1210 |
155.00 |
|
|
|
| 30 |
B |
1207 |
1153 |
140.15 |
|
|
|
| 31 |
B |
1069 |
1020 |
4.55 |
|
|
|
| 32 |
B |
1004 |
959 |
0.87 |
|
|
|
| 33 |
B |
907 |
866 |
16.10 |
|
|
|
| 34 |
B |
822 |
785 |
8.46 |
|
|
|
| 35 |
B |
544 |
519 |
27.07 |
|
|
|
| 36 |
B |
498 |
476 |
1.11 |
|
|
|
| 37 |
B |
402 |
384 |
1.12 |
|
|
|
| 38 |
B |
173 |
166 |
0.61 |
|
|
|
| 39 |
B |
27 |
26 |
11.26 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 27097.3 cm
-1
Scaled (by 0.9548) Zero Point Vibrational Energy (zpe) 25872.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.467 |
|
|
|
| 2 |
C |
0.432 |
|
|
|
| 3 |
C |
0.432 |
|
|
|
| 4 |
C |
-0.579 |
|
|
|
| 5 |
C |
-0.579 |
|
|
|
| 6 |
O |
-0.425 |
|
|
|
| 7 |
O |
-0.425 |
|
|
|
| 8 |
H |
0.199 |
|
|
|
| 9 |
H |
0.199 |
|
|
|
| 10 |
H |
0.201 |
|
|
|
| 11 |
H |
0.211 |
|
|
|
| 12 |
H |
0.193 |
|
|
|
| 13 |
H |
0.201 |
|
|
|
| 14 |
H |
0.211 |
|
|
|
| 15 |
H |
0.193 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.537 |
1.537 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.910 |
-9.017 |
0.000 |
| y |
-9.017 |
-44.831 |
0.000 |
| z |
0.000 |
0.000 |
-39.603 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.693 |
-9.017 |
0.000 |
| y |
-9.017 |
-3.574 |
0.000 |
| z |
0.000 |
0.000 |
4.267 |
|
| Polar |
|---|
| 3z2-r2 | 8.535 |
|---|
| x2-y2 | 1.921 |
|---|
| xy | -9.017 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.953 |
-0.229 |
0.000 |
| y |
-0.229 |
8.964 |
0.000 |
| z |
0.000 |
0.000 |
6.761 |
<r2> (average value of r
2) Å
2
| <r2> |
227.254 |
| (<r2>)1/2 |
15.075 |