Jump to
S1C2
Energy calculated at B1B95/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -345.174425 |
| Energy at 298.15K | -345.191545 |
| Nuclear repulsion energy | 426.906986 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3065 |
2946 |
0.00 |
|
|
|
| 2 |
A1' |
1468 |
1411 |
0.00 |
|
|
|
| 3 |
A1' |
1357 |
1304 |
0.00 |
|
|
|
| 4 |
A1' |
1003 |
964 |
0.00 |
|
|
|
| 5 |
A1' |
843 |
811 |
0.00 |
|
|
|
| 6 |
A1' |
617 |
593 |
0.00 |
|
|
|
| 7 |
A1" |
3096 |
2975 |
0.00 |
|
|
|
| 8 |
A1" |
1256 |
1207 |
0.00 |
|
|
|
| 9 |
A1" |
1013 |
973 |
0.00 |
|
|
|
| 10 |
A1" |
81 |
78 |
0.00 |
|
|
|
| 11 |
A2' |
3116 |
2995 |
0.00 |
|
|
|
| 12 |
A2' |
1190 |
1144 |
0.00 |
|
|
|
| 13 |
A2' |
793 |
763 |
0.00 |
|
|
|
| 14 |
A2" |
3051 |
2932 |
122.37 |
|
|
|
| 15 |
A2" |
1459 |
1402 |
7.45 |
|
|
|
| 16 |
A2" |
1375 |
1322 |
8.40 |
|
|
|
| 17 |
A2" |
1001 |
962 |
20.13 |
|
|
|
| 18 |
A2" |
798 |
767 |
65.38 |
|
|
|
| 19 |
E' |
3120 |
2999 |
83.57 |
|
|
|
| 19 |
E' |
3120 |
2999 |
83.46 |
|
|
|
| 20 |
E' |
3059 |
2941 |
117.61 |
|
|
|
| 20 |
E' |
3059 |
2941 |
117.26 |
|
|
|
| 21 |
E' |
1461 |
1404 |
10.35 |
|
|
|
| 21 |
E' |
1461 |
1404 |
10.44 |
|
|
|
| 22 |
E' |
1359 |
1306 |
14.65 |
|
|
|
| 22 |
E' |
1359 |
1306 |
14.64 |
|
|
|
| 23 |
E' |
1322 |
1271 |
0.08 |
|
|
|
| 23 |
E' |
1322 |
1271 |
0.08 |
|
|
|
| 24 |
E' |
1125 |
1081 |
39.46 |
|
|
|
| 24 |
E' |
1124 |
1081 |
39.47 |
|
|
|
| 25 |
E' |
929 |
893 |
4.23 |
|
|
|
| 25 |
E' |
929 |
893 |
4.24 |
|
|
|
| 26 |
E' |
817 |
785 |
3.93 |
|
|
|
| 26 |
E' |
817 |
785 |
3.92 |
|
|
|
| 27 |
E' |
408 |
392 |
0.00 |
|
|
|
| 27 |
E' |
408 |
392 |
0.00 |
|
|
|
| 28 |
E" |
3096 |
2976 |
0.00 |
|
|
|
| 28 |
E" |
3096 |
2976 |
0.00 |
|
|
|
| 29 |
E" |
3050 |
2932 |
0.00 |
|
|
|
| 29 |
E" |
3050 |
2932 |
0.00 |
|
|
|
| 30 |
E" |
1448 |
1392 |
0.00 |
|
|
|
| 30 |
E" |
1448 |
1392 |
0.00 |
|
|
|
| 31 |
E" |
1353 |
1300 |
0.00 |
|
|
|
| 31 |
E" |
1353 |
1300 |
0.00 |
|
|
|
| 32 |
E" |
1314 |
1263 |
0.00 |
|
|
|
| 32 |
E" |
1314 |
1263 |
0.00 |
|
|
|
| 33 |
E" |
1205 |
1158 |
0.00 |
|
|
|
| 33 |
E" |
1205 |
1158 |
0.00 |
|
|
|
| 34 |
E" |
1067 |
1026 |
0.00 |
|
|
|
| 34 |
E" |
1067 |
1026 |
0.00 |
|
|
|
| 35 |
E" |
583 |
560 |
0.00 |
|
|
|
| 35 |
E" |
583 |
560 |
0.00 |
|
|
|
| 36 |
E" |
324 |
312 |
0.00 |
|
|
|
| 36 |
E" |
324 |
312 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40327.4 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 38762.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVDZ
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.274 |
| N2 |
0.000 |
0.000 |
-1.274 |
| C3 |
0.000 |
1.371 |
0.775 |
| C4 |
1.187 |
-0.685 |
0.775 |
| C5 |
-1.187 |
-0.685 |
0.775 |
| C6 |
0.000 |
1.371 |
-0.775 |
| C7 |
-1.187 |
-0.685 |
-0.775 |
| C8 |
1.187 |
-0.685 |
-0.775 |
| H9 |
0.885 |
1.882 |
1.181 |
| H10 |
-0.885 |
1.882 |
1.181 |
| H11 |
1.188 |
-1.707 |
1.181 |
| H12 |
2.072 |
-0.175 |
1.181 |
| H13 |
-2.072 |
-0.175 |
1.181 |
| H14 |
-1.188 |
-1.707 |
1.181 |
| H15 |
-0.885 |
1.882 |
-1.181 |
| H16 |
0.885 |
1.882 |
-1.181 |
| H17 |
-1.188 |
-1.707 |
-1.181 |
| H18 |
-2.072 |
-0.175 |
-1.181 |
| H19 |
2.072 |
-0.175 |
-1.181 |
| H20 |
1.188 |
-1.707 |
-1.181 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5485 | 1.4589 | 1.4589 | 1.4589 | 2.4658 | 2.4658 | 2.4658 | 2.0818 | 2.0818 | 2.0818 | 2.0818 | 2.0818 | 2.0818 | 3.2178 | 3.2178 | 3.2178 | 3.2178 | 3.2178 | 3.2178 |
N2 | 2.5485 | | 2.4658 | 2.4658 | 2.4658 | 1.4589 | 1.4589 | 1.4589 | 3.2178 | 3.2178 | 3.2178 | 3.2178 | 3.2178 | 3.2178 | 2.0818 | 2.0818 | 2.0818 | 2.0818 | 2.0818 | 2.0818 | C3 | 1.4589 | 2.4658 | | 2.3746 | 2.3746 | 1.5507 | 2.8361 | 2.8361 | 1.0994 | 1.0994 | 3.3243 | 2.6170 | 2.6170 | 3.3243 | 2.2072 | 2.2072 | 3.8359 | 3.2422 | 3.2422 | 3.8359 | C4 | 1.4589 | 2.4658 | 2.3746 | | 2.3746 | 2.8361 | 2.8361 | 1.5507 | 2.6170 | 3.3243 | 1.0994 | 1.0994 | 3.3243 | 2.6170 | 3.8359 | 3.2422 | 3.2422 | 3.8359 | 2.2072 | 2.2072 | C5 | 1.4589 | 2.4658 | 2.3746 | 2.3746 | | 2.8361 | 1.5507 | 2.8361 | 3.3243 | 2.6170 | 2.6170 | 3.3243 | 1.0994 | 1.0994 | 3.2422 | 3.8359 | 2.2072 | 2.2072 | 3.8359 | 3.2422 | C6 | 2.4658 | 1.4589 | 1.5507 | 2.8361 | 2.8361 | | 2.3746 | 2.3746 | 2.2072 | 2.2072 | 3.8359 | 3.2422 | 3.2422 | 3.8359 | 1.0994 | 1.0994 | 3.3243 | 2.6170 | 2.6170 | 3.3243 | C7 | 2.4658 | 1.4589 | 2.8361 | 2.8361 | 1.5507 | 2.3746 | | 2.3746 | 3.8359 | 3.2422 | 3.2422 | 3.8359 | 2.2072 | 2.2072 | 2.6170 | 3.3243 | 1.0994 | 1.0994 | 3.3243 | 2.6170 | C8 | 2.4658 | 1.4589 | 2.8361 | 1.5507 | 2.8361 | 2.3746 | 2.3746 | | 3.2422 | 3.8359 | 2.2072 | 2.2072 | 3.8359 | 3.2422 | 3.3243 | 2.6170 | 2.6170 | 3.3243 | 1.0994 | 1.0994 | H9 | 2.0818 | 3.2178 | 1.0994 | 2.6170 | 3.3243 | 2.2072 | 3.8359 | 3.2422 | | 1.7696 | 3.6022 | 2.3751 | 3.6022 | 4.1447 | 2.9515 | 2.3623 | 4.7706 | 4.3076 | 3.3499 | 4.3076 | H10 | 2.0818 | 3.2178 | 1.0994 | 3.3243 | 2.6170 | 2.2072 | 3.2422 | 3.8359 | 1.7696 | | 4.1447 | 3.6022 | 2.3751 | 3.6022 | 2.3623 | 2.9515 | 4.3076 | 3.3499 | 4.3076 | 4.7706 | H11 | 2.0818 | 3.2178 | 3.3243 | 1.0994 | 2.6170 | 3.8359 | 3.2422 | 2.2072 | 3.6022 | 4.1447 | | 1.7696 | 3.6022 | 2.3751 | 4.7706 | 4.3076 | 3.3499 | 4.3076 | 2.9516 | 2.3623 | H12 | 2.0818 | 3.2178 | 2.6170 | 1.0994 | 3.3243 | 3.2422 | 3.8359 | 2.2072 | 2.3751 | 3.6022 | 1.7696 | | 4.1447 | 3.6022 | 4.3076 | 3.3499 | 4.3076 | 4.7706 | 2.3623 | 2.9516 | H13 | 2.0818 | 3.2178 | 2.6170 | 3.3243 | 1.0994 | 3.2422 | 2.2072 | 3.8359 | 3.6022 | 2.3751 | 3.6022 | 4.1447 | | 1.7696 | 3.3499 | 4.3076 | 2.9516 | 2.3623 | 4.7706 | 4.3076 | H14 | 2.0818 | 3.2178 | 3.3243 | 2.6170 | 1.0994 | 3.8359 | 2.2072 | 3.2422 | 4.1447 | 3.6022 | 2.3751 | 3.6022 | 1.7696 | | 4.3076 | 4.7706 | 2.3623 | 2.9516 | 4.3076 | 3.3499 | H15 | 3.2178 | 2.0818 | 2.2072 | 3.8359 | 3.2422 | 1.0994 | 2.6170 | 3.3243 | 2.9515 | 2.3623 | 4.7706 | 4.3076 | 3.3499 | 4.3076 | | 1.7696 | 3.6022 | 2.3751 | 3.6022 | 4.1447 | H16 | 3.2178 | 2.0818 | 2.2072 | 3.2422 | 3.8359 | 1.0994 | 3.3243 | 2.6170 | 2.3623 | 2.9515 | 4.3076 | 3.3499 | 4.3076 | 4.7706 | 1.7696 | | 4.1447 | 3.6022 | 2.3751 | 3.6022 | H17 | 3.2178 | 2.0818 | 3.8359 | 3.2422 | 2.2072 | 3.3243 | 1.0994 | 2.6170 | 4.7706 | 4.3076 | 3.3499 | 4.3076 | 2.9516 | 2.3623 | 3.6022 | 4.1447 | | 1.7696 | 3.6022 | 2.3751 | H18 | 3.2178 | 2.0818 | 3.2422 | 3.8359 | 2.2072 | 2.6170 | 1.0994 | 3.3243 | 4.3076 | 3.3499 | 4.3076 | 4.7706 | 2.3623 | 2.9516 | 2.3751 | 3.6022 | 1.7696 | | 4.1447 | 3.6022 | H19 | 3.2178 | 2.0818 | 3.2422 | 2.2072 | 3.8359 | 2.6170 | 3.3243 | 1.0994 | 3.3499 | 4.3076 | 2.9516 | 2.3623 | 4.7706 | 4.3076 | 3.6022 | 2.3751 | 3.6022 | 4.1447 | | 1.7696 | H20 | 3.2178 | 2.0818 | 3.8359 | 2.2072 | 3.2422 | 3.3243 | 2.6170 | 1.0994 | 4.3076 | 4.7706 | 2.3623 | 2.9516 | 4.3076 | 3.3499 | 4.1447 | 3.6022 | 2.3751 | 3.6022 | 1.7696 | |
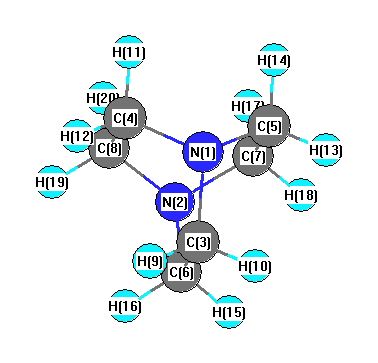 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.996 |
|
N1 |
C3 |
H9 |
108.098 |
| N1 |
C3 |
H10 |
108.098 |
|
N1 |
C4 |
C8 |
109.996 |
| N1 |
C4 |
H11 |
108.098 |
|
N1 |
C4 |
H12 |
108.098 |
| N1 |
C5 |
C7 |
109.996 |
|
N1 |
C5 |
H13 |
108.098 |
| N1 |
C5 |
H14 |
108.098 |
|
N2 |
C6 |
C3 |
109.996 |
| N2 |
C6 |
H15 |
108.098 |
|
N2 |
C6 |
H16 |
108.098 |
| N2 |
C7 |
C5 |
109.996 |
|
N2 |
C7 |
H17 |
108.098 |
| N2 |
C7 |
H18 |
108.098 |
|
N2 |
C8 |
C4 |
109.996 |
| N2 |
C8 |
H19 |
108.098 |
|
N2 |
C8 |
H20 |
108.098 |
| C3 |
N1 |
C4 |
108.941 |
|
C3 |
N1 |
C5 |
108.941 |
| C3 |
C6 |
H15 |
111.661 |
|
C3 |
C6 |
H16 |
111.661 |
| C4 |
N1 |
C5 |
108.941 |
|
C4 |
C8 |
H19 |
111.661 |
| C4 |
C8 |
H20 |
111.661 |
|
C5 |
C6 |
H15 |
101.654 |
| C5 |
C6 |
H16 |
151.142 |
|
C6 |
N2 |
C7 |
108.941 |
| C6 |
N2 |
C8 |
108.941 |
|
C6 |
C3 |
H9 |
111.661 |
| C6 |
C3 |
H10 |
111.661 |
|
C7 |
N2 |
C8 |
108.941 |
| C7 |
C5 |
H13 |
111.661 |
|
C7 |
C5 |
H14 |
111.661 |
| C8 |
C4 |
H11 |
111.661 |
|
C8 |
C4 |
H12 |
111.661 |
| H9 |
C3 |
H10 |
107.174 |
|
H11 |
C4 |
H12 |
107.174 |
| H13 |
C5 |
H14 |
107.174 |
|
H15 |
C6 |
H16 |
107.174 |
| H17 |
C7 |
H18 |
107.174 |
|
H19 |
C8 |
H20 |
107.174 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.291 |
|
|
|
| 2 |
N |
-0.291 |
|
|
|
| 3 |
C |
0.059 |
|
|
|
| 4 |
C |
0.059 |
|
|
|
| 5 |
C |
0.059 |
|
|
|
| 6 |
C |
0.059 |
|
|
|
| 7 |
C |
0.059 |
|
|
|
| 8 |
C |
0.059 |
|
|
|
| 9 |
H |
0.019 |
|
|
|
| 10 |
H |
0.019 |
|
|
|
| 11 |
H |
0.019 |
|
|
|
| 12 |
H |
0.019 |
|
|
|
| 13 |
H |
0.019 |
|
|
|
| 14 |
H |
0.019 |
|
|
|
| 15 |
H |
0.019 |
|
|
|
| 16 |
H |
0.019 |
|
|
|
| 17 |
H |
0.019 |
|
|
|
| 18 |
H |
0.019 |
|
|
|
| 19 |
H |
0.019 |
|
|
|
| 20 |
H |
0.019 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.924 |
0.000 |
0.000 |
| y |
0.000 |
-46.924 |
0.000 |
| z |
0.000 |
0.000 |
-56.702 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.889 |
0.000 |
0.000 |
| y |
0.000 |
4.889 |
0.000 |
| z |
0.000 |
0.000 |
-9.778 |
|
| Polar |
|---|
| 3z2-r2 | -19.556 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.412 |
0.000 |
0.000 |
| y |
0.000 |
11.410 |
0.000 |
| z |
0.000 |
0.000 |
9.851 |
<r2> (average value of r
2) Å
2
| <r2> |
212.025 |
| (<r2>)1/2 |
14.561 |
Jump to
S1C1
Energy calculated at B1B95/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -345.174415 |
| Energy at 298.15K | |
| HF Energy | -345.174415 |
| Nuclear repulsion energy | 426.898668 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVDZ
Geometric Data calculated at B1B95/cc-pVDZ
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.275 |
| N2 |
0.000 |
0.000 |
-1.275 |
| C3 |
-0.003 |
1.371 |
0.775 |
| C4 |
1.189 |
-0.683 |
0.775 |
| C5 |
-1.186 |
-0.688 |
0.775 |
| C6 |
0.003 |
1.371 |
-0.775 |
| C7 |
-1.189 |
-0.683 |
-0.775 |
| C8 |
1.186 |
-0.688 |
-0.775 |
| H9 |
0.878 |
1.886 |
1.185 |
| H10 |
-0.892 |
1.878 |
1.177 |
| H11 |
1.195 |
-1.703 |
1.185 |
| H12 |
2.073 |
-0.167 |
1.177 |
| H13 |
-2.072 |
-0.183 |
1.185 |
| H14 |
-1.181 |
-1.711 |
1.177 |
| H15 |
-0.878 |
1.886 |
-1.185 |
| H16 |
0.892 |
1.878 |
-1.177 |
| H17 |
-1.195 |
-1.703 |
-1.185 |
| H18 |
-2.073 |
-0.167 |
-1.177 |
| H19 |
2.072 |
-0.183 |
-1.185 |
| H20 |
1.181 |
-1.711 |
-1.177 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5490 | 1.4590 | 1.4590 | 1.4590 | 2.4660 | 2.4660 | 2.4660 | 2.0822 | 2.0815 | 2.0822 | 2.0815 | 2.0822 | 2.0815 | 3.2211 | 3.2148 | 3.2211 | 3.2148 | 3.2211 | 3.2148 |
N2 | 2.5490 | | 2.4660 | 2.4660 | 2.4660 | 1.4590 | 1.4590 | 1.4590 | 3.2211 | 3.2148 | 3.2211 | 3.2148 | 3.2211 | 3.2148 | 2.0822 | 2.0815 | 2.0822 | 2.0815 | 2.0822 | 2.0815 | C3 | 1.4590 | 2.4660 | | 2.3746 | 2.3746 | 1.5506 | 2.8334 | 2.8386 | 1.0994 | 1.0994 | 3.3245 | 2.6144 | 2.6199 | 3.3241 | 2.2072 | 2.2071 | 3.8355 | 3.2343 | 3.2501 | 3.8361 | C4 | 1.4590 | 2.4660 | 2.3746 | | 2.3746 | 2.8334 | 2.8386 | 1.5506 | 2.6199 | 3.3241 | 1.0994 | 1.0994 | 3.3245 | 2.6144 | 3.8355 | 3.2343 | 3.2501 | 3.8361 | 2.2072 | 2.2071 | C5 | 1.4590 | 2.4660 | 2.3746 | 2.3746 | | 2.8386 | 1.5506 | 2.8334 | 3.3245 | 2.6144 | 2.6199 | 3.3241 | 1.0994 | 1.0994 | 3.2501 | 3.8361 | 2.2072 | 2.2071 | 3.8355 | 3.2343 | C6 | 2.4660 | 1.4590 | 1.5506 | 2.8334 | 2.8386 | | 2.3746 | 2.3746 | 2.2072 | 2.2071 | 3.8355 | 3.2343 | 3.2501 | 3.8361 | 1.0994 | 1.0994 | 3.3245 | 2.6144 | 2.6199 | 3.3241 | C7 | 2.4660 | 1.4590 | 2.8334 | 2.8386 | 1.5506 | 2.3746 | | 2.3746 | 3.8355 | 3.2343 | 3.2501 | 3.8361 | 2.2072 | 2.2071 | 2.6199 | 3.3241 | 1.0994 | 1.0994 | 3.3245 | 2.6144 | C8 | 2.4660 | 1.4590 | 2.8386 | 1.5506 | 2.8334 | 2.3746 | 2.3746 | | 3.2501 | 3.8361 | 2.2072 | 2.2071 | 3.8355 | 3.2343 | 3.3245 | 2.6144 | 2.6199 | 3.3241 | 1.0994 | 1.0994 | H9 | 2.0822 | 3.2211 | 1.0994 | 2.6199 | 3.3245 | 2.2072 | 3.8355 | 3.2501 | | 1.7694 | 3.6032 | 2.3754 | 3.6032 | 4.1448 | 2.9487 | 2.3622 | 4.7739 | 4.3009 | 3.3648 | 4.3143 | H10 | 2.0815 | 3.2148 | 1.0994 | 3.3241 | 2.6144 | 2.2071 | 3.2343 | 3.8361 | 1.7694 | | 4.1448 | 3.6013 | 2.3754 | 3.6013 | 2.3622 | 2.9540 | 4.3009 | 3.3351 | 4.3143 | 4.7673 | H11 | 2.0822 | 3.2211 | 3.3245 | 1.0994 | 2.6199 | 3.8355 | 3.2501 | 2.2072 | 3.6032 | 4.1448 | | 1.7694 | 3.6032 | 2.3754 | 4.7739 | 4.3009 | 3.3648 | 4.3143 | 2.9487 | 2.3622 | H12 | 2.0815 | 3.2148 | 2.6144 | 1.0994 | 3.3241 | 3.2343 | 3.8361 | 2.2071 | 2.3754 | 3.6013 | 1.7694 | | 4.1448 | 3.6013 | 4.3009 | 3.3351 | 4.3143 | 4.7673 | 2.3622 | 2.9540 | H13 | 2.0822 | 3.2211 | 2.6199 | 3.3245 | 1.0994 | 3.2501 | 2.2072 | 3.8355 | 3.6032 | 2.3754 | 3.6032 | 4.1448 | | 1.7694 | 3.3648 | 4.3143 | 2.9487 | 2.3622 | 4.7739 | 4.3009 | H14 | 2.0815 | 3.2148 | 3.3241 | 2.6144 | 1.0994 | 3.8361 | 2.2071 | 3.2343 | 4.1448 | 3.6013 | 2.3754 | 3.6013 | 1.7694 | | 4.3143 | 4.7673 | 2.3622 | 2.9540 | 4.3009 | 3.3351 | H15 | 3.2211 | 2.0822 | 2.2072 | 3.8355 | 3.2501 | 1.0994 | 2.6199 | 3.3245 | 2.9487 | 2.3622 | 4.7739 | 4.3009 | 3.3648 | 4.3143 | | 1.7694 | 3.6032 | 2.3754 | 3.6032 | 4.1448 | H16 | 3.2148 | 2.0815 | 2.2071 | 3.2343 | 3.8361 | 1.0994 | 3.3241 | 2.6144 | 2.3622 | 2.9540 | 4.3009 | 3.3351 | 4.3143 | 4.7673 | 1.7694 | | 4.1448 | 3.6013 | 2.3754 | 3.6013 | H17 | 3.2211 | 2.0822 | 3.8355 | 3.2501 | 2.2072 | 3.3245 | 1.0994 | 2.6199 | 4.7739 | 4.3009 | 3.3648 | 4.3143 | 2.9487 | 2.3622 | 3.6032 | 4.1448 | | 1.7694 | 3.6032 | 2.3754 | H18 | 3.2148 | 2.0815 | 3.2343 | 3.8361 | 2.2071 | 2.6144 | 1.0994 | 3.3241 | 4.3009 | 3.3351 | 4.3143 | 4.7673 | 2.3622 | 2.9540 | 2.3754 | 3.6013 | 1.7694 | | 4.1448 | 3.6013 | H19 | 3.2211 | 2.0822 | 3.2501 | 2.2072 | 3.8355 | 2.6199 | 3.3245 | 1.0994 | 3.3648 | 4.3143 | 2.9487 | 2.3622 | 4.7739 | 4.3009 | 3.6032 | 2.3754 | 3.6032 | 4.1448 | | 1.7694 | H20 | 3.2148 | 2.0815 | 3.8361 | 2.2071 | 3.2343 | 3.3241 | 2.6144 | 1.0994 | 4.3143 | 4.7673 | 2.3622 | 2.9540 | 4.3009 | 3.3351 | 4.1448 | 3.6013 | 2.3754 | 3.6013 | 1.7694 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
110.008 |
|
N1 |
C3 |
H9 |
108.130 |
| N1 |
C3 |
H10 |
108.070 |
|
N1 |
C4 |
C8 |
110.008 |
| N1 |
C4 |
H11 |
108.130 |
|
N1 |
C4 |
H12 |
108.070 |
| N1 |
C5 |
C7 |
110.008 |
|
N1 |
C5 |
H13 |
108.130 |
| N1 |
C5 |
H14 |
108.070 |
|
N2 |
C6 |
C3 |
110.008 |
| N2 |
C6 |
H15 |
108.130 |
|
N2 |
C6 |
H16 |
108.070 |
| N2 |
C7 |
C5 |
110.008 |
|
N2 |
C7 |
H17 |
108.130 |
| N2 |
C7 |
H18 |
108.070 |
|
N2 |
C8 |
C4 |
110.008 |
| N2 |
C8 |
H19 |
108.130 |
|
N2 |
C8 |
H20 |
108.070 |
| C3 |
N1 |
C4 |
108.929 |
|
C3 |
N1 |
C5 |
108.929 |
| C3 |
C6 |
H15 |
111.662 |
|
C3 |
C6 |
H16 |
111.655 |
| C4 |
N1 |
C5 |
108.929 |
|
C4 |
C8 |
H19 |
111.662 |
| C4 |
C8 |
H20 |
111.655 |
|
C5 |
C6 |
H15 |
101.992 |
| C5 |
C6 |
H16 |
150.816 |
|
C6 |
N2 |
C7 |
108.929 |
| C6 |
N2 |
C8 |
108.929 |
|
C6 |
C3 |
H9 |
111.662 |
| C6 |
C3 |
H10 |
111.655 |
|
C7 |
N2 |
C8 |
108.929 |
| C7 |
C5 |
H13 |
111.662 |
|
C7 |
C5 |
H14 |
111.655 |
| C8 |
C4 |
H11 |
111.662 |
|
C8 |
C4 |
H12 |
111.655 |
| H9 |
C3 |
H10 |
107.162 |
|
H11 |
C4 |
H12 |
107.162 |
| H13 |
C5 |
H14 |
107.162 |
|
H15 |
C6 |
H16 |
107.162 |
| H17 |
C7 |
H18 |
107.162 |
|
H19 |
C8 |
H20 |
107.162 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.291 |
|
|
|
| 2 |
N |
-0.291 |
|
|
|
| 3 |
C |
0.059 |
|
|
|
| 4 |
C |
0.059 |
|
|
|
| 5 |
C |
0.059 |
|
|
|
| 6 |
C |
0.059 |
|
|
|
| 7 |
C |
0.059 |
|
|
|
| 8 |
C |
0.059 |
|
|
|
| 9 |
H |
0.019 |
|
|
|
| 10 |
H |
0.019 |
|
|
|
| 11 |
H |
0.019 |
|
|
|
| 12 |
H |
0.019 |
|
|
|
| 13 |
H |
0.019 |
|
|
|
| 14 |
H |
0.019 |
|
|
|
| 15 |
H |
0.019 |
|
|
|
| 16 |
H |
0.019 |
|
|
|
| 17 |
H |
0.019 |
|
|
|
| 18 |
H |
0.019 |
|
|
|
| 19 |
H |
0.019 |
|
|
|
| 20 |
H |
0.019 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.923 |
0.000 |
0.000 |
| y |
0.000 |
-46.923 |
0.000 |
| z |
0.000 |
0.000 |
-56.705 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.891 |
0.000 |
0.000 |
| y |
0.000 |
4.891 |
0.000 |
| z |
0.000 |
0.000 |
-9.782 |
|
| Polar |
|---|
| 3z2-r2 | -19.564 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.411 |
0.000 |
0.000 |
| y |
0.000 |
11.409 |
0.000 |
| z |
0.000 |
0.000 |
9.851 |
<r2> (average value of r
2) Å
2
| <r2> |
212.031 |
| (<r2>)1/2 |
14.561 |