Jump to
S1C2
S2C1
S2C2
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.408164 |
| Energy at 298.15K | -131.407362 |
| HF Energy | -131.408164 |
| Nuclear repulsion energy | 47.684881 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3416 |
3270 |
46.68 |
|
|
|
| 2 |
A' |
1770 |
1694 |
38.86 |
|
|
|
| 3 |
A' |
1257 |
1203 |
7.14 |
|
|
|
| 4 |
A' |
449 |
430 |
4.04 |
|
|
|
| 5 |
A' |
295 |
282 |
43.74 |
|
|
|
| 6 |
A" |
446 |
427 |
0.79 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3817.0 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 3653.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.086 |
-1.205 |
0.000 |
| C2 |
0.000 |
0.085 |
0.000 |
| N3 |
-0.151 |
1.270 |
0.000 |
| H4 |
0.547 |
-2.165 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.2931 | 2.4867 | 1.0644 |
C2 | 1.2931 | | 1.1947 | 2.3151 | N3 | 2.4867 | 1.1947 | | 3.5049 | H4 | 1.0644 | 2.3151 | 3.5049 | |
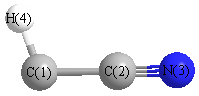 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
176.513 |
|
C2 |
C1 |
H4 |
158.133 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.407972 |
| Energy at 298.15K | |
| HF Energy | -131.407972 |
| Nuclear repulsion energy | 47.722027 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3454 |
3306 |
66.15 |
|
|
|
| 2 |
Σ |
1726 |
1652 |
40.90 |
|
|
|
| 3 |
Σ |
1284 |
1229 |
5.06 |
|
|
|
| 4 |
Π |
445 |
426 |
0.19 |
|
|
|
| 4 |
Π |
445 |
426 |
0.19 |
|
|
|
| 5 |
Π |
207i |
198i |
47.23 |
|
|
|
| 5 |
Π |
207i |
198i |
47.23 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3470.5 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 3321.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.200 |
| C2 |
0.000 |
0.000 |
0.081 |
| N3 |
0.000 |
0.000 |
1.282 |
| H4 |
0.000 |
0.000 |
-2.262 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.2815 | 2.4822 | 1.0614 |
C2 | 1.2815 | | 1.2007 | 2.3429 | N3 | 2.4822 | 1.2007 | | 3.5436 | H4 | 1.0614 | 2.3429 | 3.5436 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.155 |
|
|
|
| 2 |
C |
0.053 |
|
|
|
| 3 |
N |
-0.077 |
|
|
|
| 4 |
H |
0.179 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.232 |
3.232 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-17.207 |
0.000 |
0.000 |
| y |
0.000 |
-17.207 |
0.000 |
| z |
0.000 |
0.000 |
-15.377 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.915 |
0.000 |
0.000 |
| y |
0.000 |
-0.915 |
0.000 |
| z |
0.000 |
0.000 |
1.830 |
|
| Polar |
|---|
| 3z2-r2 | 3.660 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.399 |
0.000 |
0.000 |
| y |
0.000 |
2.399 |
0.000 |
| z |
0.000 |
0.000 |
6.637 |
<r2> (average value of r
2) Å
2
| <r2> |
35.668 |
| (<r2>)1/2 |
5.972 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.382239 |
| Energy at 298.15K | -131.381691 |
| HF Energy | -131.382239 |
| Nuclear repulsion energy | 47.324871 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3084 |
2951 |
4.48 |
|
|
|
| 2 |
A' |
2121 |
2030 |
24.13 |
|
|
|
| 3 |
A' |
1096 |
1049 |
39.78 |
|
|
|
| 4 |
A' |
946 |
905 |
42.00 |
|
|
|
| 5 |
A' |
460 |
440 |
24.29 |
|
|
|
| 6 |
A" |
310 |
297 |
11.88 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4007.7 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 3835.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.042 |
-1.275 |
0.000 |
| C2 |
0.000 |
0.092 |
0.000 |
| N3 |
-0.190 |
1.247 |
0.000 |
| H4 |
1.076 |
-1.637 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3674 | 2.5326 | 1.0955 |
C2 | 1.3674 | | 1.1707 | 2.0366 | N3 | 2.5326 | 1.1707 | | 3.1499 | H4 | 1.0955 | 2.0366 | 3.1499 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.149 |
|
|
|
| 2 |
C |
0.006 |
|
|
|
| 3 |
N |
-0.018 |
|
|
|
| 4 |
H |
0.161 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.672 |
-1.424 |
0.000 |
2.196 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-17.044 |
-2.471 |
0.000 |
| y |
-2.471 |
-20.740 |
0.000 |
| z |
0.000 |
0.000 |
-15.874 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.263 |
-2.471 |
0.000 |
| y |
-2.471 |
-4.281 |
0.000 |
| z |
0.000 |
0.000 |
3.018 |
|
| Polar |
|---|
| 3z2-r2 | 6.036 |
|---|
| x2-y2 | 3.696 |
|---|
| xy | -2.471 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.961 |
-0.518 |
0.000 |
| y |
-0.518 |
6.557 |
0.000 |
| z |
0.000 |
0.000 |
2.763 |
<r2> (average value of r
2) Å
2
| <r2> |
35.962 |
| (<r2>)1/2 |
5.997 |
Jump to
S1C1
S1C2
S2C1
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.382239 |
| Energy at 298.15K | -131.381691 |
| HF Energy | -131.382239 |
| Nuclear repulsion energy | 47.324871 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability