Jump to
S1C2
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -345.267560 |
| Energy at 298.15K | -345.284572 |
| Nuclear repulsion energy | 428.236304 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3060 |
2928 |
0.00 |
|
|
|
| 2 |
A1' |
1493 |
1429 |
0.00 |
|
|
|
| 3 |
A1' |
1359 |
1301 |
0.00 |
|
|
|
| 4 |
A1' |
992 |
950 |
0.00 |
|
|
|
| 5 |
A1' |
840 |
804 |
0.00 |
|
|
|
| 6 |
A1' |
616 |
590 |
0.00 |
|
|
|
| 7 |
A1" |
3083 |
2951 |
0.00 |
|
|
|
| 8 |
A1" |
1266 |
1212 |
0.00 |
|
|
|
| 9 |
A1" |
1019 |
975 |
0.00 |
|
|
|
| 10 |
A1" |
31 |
29 |
0.00 |
|
|
|
| 11 |
A2' |
3105 |
2972 |
0.00 |
|
|
|
| 12 |
A2' |
1197 |
1146 |
0.00 |
|
|
|
| 13 |
A2' |
803 |
768 |
0.00 |
|
|
|
| 14 |
A2" |
3046 |
2916 |
113.56 |
|
|
|
| 15 |
A2" |
1485 |
1421 |
7.62 |
|
|
|
| 16 |
A2" |
1385 |
1326 |
3.51 |
|
|
|
| 17 |
A2" |
1002 |
959 |
22.14 |
|
|
|
| 18 |
A2" |
791 |
757 |
66.06 |
|
|
|
| 19 |
E' |
3111 |
2977 |
90.39 |
|
|
|
| 19 |
E' |
3111 |
2977 |
90.44 |
|
|
|
| 20 |
E' |
3054 |
2923 |
121.98 |
|
|
|
| 20 |
E' |
3054 |
2923 |
121.72 |
|
|
|
| 21 |
E' |
1488 |
1424 |
10.67 |
|
|
|
| 21 |
E' |
1488 |
1424 |
10.73 |
|
|
|
| 22 |
E' |
1362 |
1304 |
14.41 |
|
|
|
| 22 |
E' |
1362 |
1304 |
14.42 |
|
|
|
| 23 |
E' |
1325 |
1269 |
0.34 |
|
|
|
| 23 |
E' |
1325 |
1269 |
0.34 |
|
|
|
| 24 |
E' |
1114 |
1067 |
42.76 |
|
|
|
| 24 |
E' |
1114 |
1067 |
42.79 |
|
|
|
| 25 |
E' |
922 |
883 |
4.27 |
|
|
|
| 25 |
E' |
922 |
882 |
4.28 |
|
|
|
| 26 |
E' |
822 |
787 |
3.45 |
|
|
|
| 26 |
E' |
822 |
787 |
3.44 |
|
|
|
| 27 |
E' |
403 |
386 |
0.00 |
|
|
|
| 27 |
E' |
403 |
386 |
0.00 |
|
|
|
| 28 |
E" |
3085 |
2952 |
0.00 |
|
|
|
| 28 |
E" |
3085 |
2952 |
0.00 |
|
|
|
| 29 |
E" |
3045 |
2914 |
0.00 |
|
|
|
| 29 |
E" |
3045 |
2914 |
0.00 |
|
|
|
| 30 |
E" |
1475 |
1412 |
0.00 |
|
|
|
| 30 |
E" |
1475 |
1412 |
0.00 |
|
|
|
| 31 |
E" |
1354 |
1296 |
0.00 |
|
|
|
| 31 |
E" |
1354 |
1296 |
0.00 |
|
|
|
| 32 |
E" |
1327 |
1270 |
0.00 |
|
|
|
| 32 |
E" |
1327 |
1270 |
0.00 |
|
|
|
| 33 |
E" |
1210 |
1158 |
0.00 |
|
|
|
| 33 |
E" |
1210 |
1158 |
0.00 |
|
|
|
| 34 |
E" |
1064 |
1018 |
0.00 |
|
|
|
| 34 |
E" |
1064 |
1018 |
0.00 |
|
|
|
| 35 |
E" |
579 |
554 |
0.00 |
|
|
|
| 35 |
E" |
579 |
554 |
0.00 |
|
|
|
| 36 |
E" |
315 |
301 |
0.00 |
|
|
|
| 36 |
E" |
315 |
301 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40340.3 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 38609.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.270 |
| N2 |
0.000 |
0.000 |
-1.270 |
| C3 |
0.000 |
1.369 |
0.774 |
| C4 |
1.185 |
-0.684 |
0.774 |
| C5 |
-1.185 |
-0.684 |
0.774 |
| C6 |
0.000 |
1.369 |
-0.774 |
| C7 |
-1.185 |
-0.684 |
-0.774 |
| C8 |
1.185 |
-0.684 |
-0.774 |
| H9 |
0.879 |
1.875 |
1.173 |
| H10 |
-0.879 |
1.875 |
1.173 |
| H11 |
1.185 |
-1.698 |
1.173 |
| H12 |
2.063 |
-0.177 |
1.173 |
| H13 |
-2.063 |
-0.177 |
1.173 |
| H14 |
-1.185 |
-1.698 |
1.173 |
| H15 |
-0.879 |
1.875 |
-1.173 |
| H16 |
0.879 |
1.875 |
-1.173 |
| H17 |
-1.185 |
-1.698 |
-1.173 |
| H18 |
-2.063 |
-0.177 |
-1.173 |
| H19 |
2.063 |
-0.177 |
-1.173 |
| H20 |
1.185 |
-1.698 |
-1.173 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5397 | 1.4558 | 1.4558 | 1.4558 | 2.4600 | 2.4600 | 2.4600 | 2.0731 | 2.0731 | 2.0731 | 2.0731 | 2.0731 | 2.0731 | 3.2028 | 3.2028 | 3.2028 | 3.2028 | 3.2028 | 3.2028 |
N2 | 2.5397 | | 2.4600 | 2.4600 | 2.4600 | 1.4558 | 1.4558 | 1.4558 | 3.2028 | 3.2028 | 3.2028 | 3.2028 | 3.2028 | 3.2028 | 2.0731 | 2.0731 | 2.0731 | 2.0731 | 2.0731 | 2.0731 | C3 | 1.4558 | 2.4600 | | 2.3709 | 2.3709 | 1.5483 | 2.8316 | 2.8316 | 1.0898 | 1.0898 | 3.3123 | 2.6087 | 2.6087 | 3.3123 | 2.1957 | 2.1957 | 3.8216 | 3.2309 | 3.2309 | 3.8216 | C4 | 1.4558 | 2.4600 | 2.3709 | | 2.3709 | 2.8316 | 2.8316 | 1.5483 | 2.6087 | 3.3123 | 1.0898 | 1.0898 | 3.3123 | 2.6087 | 3.8216 | 3.2309 | 3.2309 | 3.8216 | 2.1957 | 2.1957 | C5 | 1.4558 | 2.4600 | 2.3709 | 2.3709 | | 2.8316 | 1.5483 | 2.8316 | 3.3123 | 2.6087 | 2.6087 | 3.3123 | 1.0898 | 1.0898 | 3.2309 | 3.8216 | 2.1957 | 2.1957 | 3.8216 | 3.2309 | C6 | 2.4600 | 1.4558 | 1.5483 | 2.8316 | 2.8316 | | 2.3709 | 2.3709 | 2.1957 | 2.1957 | 3.8216 | 3.2309 | 3.2309 | 3.8216 | 1.0898 | 1.0898 | 3.3123 | 2.6087 | 2.6087 | 3.3123 | C7 | 2.4600 | 1.4558 | 2.8316 | 2.8316 | 1.5483 | 2.3709 | | 2.3709 | 3.8216 | 3.2309 | 3.2309 | 3.8216 | 2.1957 | 2.1957 | 2.6087 | 3.3123 | 1.0898 | 1.0898 | 3.3123 | 2.6087 | C8 | 2.4600 | 1.4558 | 2.8316 | 1.5483 | 2.8316 | 2.3709 | 2.3709 | | 3.2309 | 3.8216 | 2.1957 | 2.1957 | 3.8216 | 3.2309 | 3.3123 | 2.6087 | 2.6087 | 3.3123 | 1.0898 | 1.0898 | H9 | 2.0731 | 3.2028 | 1.0898 | 2.6087 | 3.3123 | 2.1957 | 3.8216 | 3.2309 | | 1.7572 | 3.5868 | 2.3693 | 3.5868 | 4.1265 | 2.9318 | 2.3468 | 4.7472 | 4.2863 | 3.3348 | 4.2863 | H10 | 2.0731 | 3.2028 | 1.0898 | 3.3123 | 2.6087 | 2.1957 | 3.2309 | 3.8216 | 1.7572 | | 4.1265 | 3.5868 | 2.3693 | 3.5868 | 2.3468 | 2.9318 | 4.2863 | 3.3348 | 4.2863 | 4.7472 | H11 | 2.0731 | 3.2028 | 3.3123 | 1.0898 | 2.6087 | 3.8216 | 3.2309 | 2.1957 | 3.5868 | 4.1265 | | 1.7572 | 3.5868 | 2.3693 | 4.7472 | 4.2863 | 3.3348 | 4.2863 | 2.9318 | 2.3468 | H12 | 2.0731 | 3.2028 | 2.6087 | 1.0898 | 3.3123 | 3.2309 | 3.8216 | 2.1957 | 2.3693 | 3.5868 | 1.7572 | | 4.1265 | 3.5868 | 4.2863 | 3.3348 | 4.2863 | 4.7472 | 2.3468 | 2.9318 | H13 | 2.0731 | 3.2028 | 2.6087 | 3.3123 | 1.0898 | 3.2309 | 2.1957 | 3.8216 | 3.5868 | 2.3693 | 3.5868 | 4.1265 | | 1.7572 | 3.3348 | 4.2863 | 2.9318 | 2.3468 | 4.7472 | 4.2863 | H14 | 2.0731 | 3.2028 | 3.3123 | 2.6087 | 1.0898 | 3.8216 | 2.1957 | 3.2309 | 4.1265 | 3.5868 | 2.3693 | 3.5868 | 1.7572 | | 4.2863 | 4.7472 | 2.3468 | 2.9318 | 4.2863 | 3.3348 | H15 | 3.2028 | 2.0731 | 2.1957 | 3.8216 | 3.2309 | 1.0898 | 2.6087 | 3.3123 | 2.9318 | 2.3468 | 4.7472 | 4.2863 | 3.3348 | 4.2863 | | 1.7572 | 3.5868 | 2.3693 | 3.5868 | 4.1265 | H16 | 3.2028 | 2.0731 | 2.1957 | 3.2309 | 3.8216 | 1.0898 | 3.3123 | 2.6087 | 2.3468 | 2.9318 | 4.2863 | 3.3348 | 4.2863 | 4.7472 | 1.7572 | | 4.1265 | 3.5868 | 2.3693 | 3.5868 | H17 | 3.2028 | 2.0731 | 3.8216 | 3.2309 | 2.1957 | 3.3123 | 1.0898 | 2.6087 | 4.7472 | 4.2863 | 3.3348 | 4.2863 | 2.9318 | 2.3468 | 3.5868 | 4.1265 | | 1.7572 | 3.5868 | 2.3693 | H18 | 3.2028 | 2.0731 | 3.2309 | 3.8216 | 2.1957 | 2.6087 | 1.0898 | 3.3123 | 4.2863 | 3.3348 | 4.2863 | 4.7472 | 2.3468 | 2.9318 | 2.3693 | 3.5868 | 1.7572 | | 4.1265 | 3.5868 | H19 | 3.2028 | 2.0731 | 3.2309 | 2.1957 | 3.8216 | 2.6087 | 3.3123 | 1.0898 | 3.3348 | 4.2863 | 2.9318 | 2.3468 | 4.7472 | 4.2863 | 3.5868 | 2.3693 | 3.5868 | 4.1265 | | 1.7572 | H20 | 3.2028 | 2.0731 | 3.8216 | 2.1957 | 3.2309 | 3.3123 | 2.6087 | 1.0898 | 4.2863 | 4.7472 | 2.3468 | 2.9318 | 4.2863 | 3.3348 | 4.1265 | 3.5868 | 2.3693 | 3.5868 | 1.7572 | |
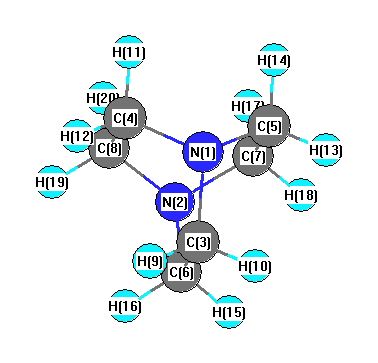 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.908 |
|
N1 |
C3 |
H9 |
108.186 |
| N1 |
C3 |
H10 |
108.186 |
|
N1 |
C4 |
C8 |
109.908 |
| N1 |
C4 |
H11 |
108.186 |
|
N1 |
C4 |
H12 |
108.186 |
| N1 |
C5 |
C7 |
109.908 |
|
N1 |
C5 |
H13 |
108.186 |
| N1 |
C5 |
H14 |
108.186 |
|
N2 |
C6 |
C3 |
109.908 |
| N2 |
C6 |
H15 |
108.186 |
|
N2 |
C6 |
H16 |
108.186 |
| N2 |
C7 |
C5 |
109.908 |
|
N2 |
C7 |
H17 |
108.186 |
| N2 |
C7 |
H18 |
108.186 |
|
N2 |
C8 |
C4 |
109.908 |
| N2 |
C8 |
H19 |
108.186 |
|
N2 |
C8 |
H20 |
108.186 |
| C3 |
N1 |
C4 |
109.031 |
|
C3 |
N1 |
C5 |
109.031 |
| C3 |
C6 |
H15 |
111.491 |
|
C3 |
C6 |
H16 |
111.491 |
| C4 |
N1 |
C5 |
109.031 |
|
C4 |
C8 |
H19 |
111.491 |
| C4 |
C8 |
H20 |
111.491 |
|
C5 |
C6 |
H15 |
101.520 |
| C5 |
C6 |
H16 |
151.010 |
|
C6 |
N2 |
C7 |
109.031 |
| C6 |
N2 |
C8 |
109.031 |
|
C6 |
C3 |
H9 |
111.491 |
| C6 |
C3 |
H10 |
111.491 |
|
C7 |
N2 |
C8 |
109.031 |
| C7 |
C5 |
H13 |
111.491 |
|
C7 |
C5 |
H14 |
111.491 |
| C8 |
C4 |
H11 |
111.491 |
|
C8 |
C4 |
H12 |
111.491 |
| H9 |
C3 |
H10 |
107.447 |
|
H11 |
C4 |
H12 |
107.447 |
| H13 |
C5 |
H14 |
107.447 |
|
H15 |
C6 |
H16 |
107.447 |
| H17 |
C7 |
H18 |
107.447 |
|
H19 |
C8 |
H20 |
107.447 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.147 |
|
|
|
| 2 |
N |
-0.147 |
|
|
|
| 3 |
C |
-0.168 |
|
|
|
| 4 |
C |
-0.168 |
|
|
|
| 5 |
C |
-0.168 |
|
|
|
| 6 |
C |
-0.168 |
|
|
|
| 7 |
C |
-0.168 |
|
|
|
| 8 |
C |
-0.168 |
|
|
|
| 9 |
H |
0.109 |
|
|
|
| 10 |
H |
0.109 |
|
|
|
| 11 |
H |
0.109 |
|
|
|
| 12 |
H |
0.109 |
|
|
|
| 13 |
H |
0.109 |
|
|
|
| 14 |
H |
0.109 |
|
|
|
| 15 |
H |
0.109 |
|
|
|
| 16 |
H |
0.109 |
|
|
|
| 17 |
H |
0.109 |
|
|
|
| 18 |
H |
0.109 |
|
|
|
| 19 |
H |
0.109 |
|
|
|
| 20 |
H |
0.109 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.936 |
0.000 |
0.000 |
| y |
0.000 |
-46.936 |
0.000 |
| z |
0.000 |
0.000 |
-57.154 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.109 |
0.000 |
0.000 |
| y |
0.000 |
5.109 |
0.000 |
| z |
0.000 |
0.000 |
-10.219 |
|
| Polar |
|---|
| 3z2-r2 | -20.437 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.012 |
0.000 |
0.000 |
| y |
0.000 |
12.011 |
0.000 |
| z |
0.000 |
0.000 |
10.925 |
<r2> (average value of r
2) Å
2
| <r2> |
211.027 |
| (<r2>)1/2 |
14.527 |
Jump to
S1C1
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -345.267567 |
| Energy at 298.15K | |
| HF Energy | -345.267567 |
| Nuclear repulsion energy | 428.283049 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.270 |
| N2 |
0.000 |
0.000 |
-1.270 |
| C3 |
-0.029 |
1.368 |
0.773 |
| C4 |
1.200 |
-0.659 |
0.773 |
| C5 |
-1.170 |
-0.709 |
0.773 |
| C6 |
0.029 |
1.368 |
-0.773 |
| C7 |
-1.200 |
-0.659 |
-0.773 |
| C8 |
1.170 |
-0.709 |
-0.773 |
| H9 |
0.812 |
1.910 |
1.205 |
| H10 |
-0.942 |
1.840 |
1.137 |
| H11 |
1.248 |
-1.658 |
1.205 |
| H12 |
2.064 |
-0.104 |
1.137 |
| H13 |
-2.060 |
-0.251 |
1.205 |
| H14 |
-1.122 |
-1.736 |
1.137 |
| H15 |
-0.812 |
1.910 |
-1.205 |
| H16 |
0.942 |
1.840 |
-1.137 |
| H17 |
-1.248 |
-1.658 |
-1.205 |
| H18 |
-2.064 |
-0.104 |
-1.137 |
| H19 |
2.060 |
-0.251 |
-1.205 |
| H20 |
1.122 |
-1.736 |
-1.137 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5405 | 1.4561 | 1.4561 | 1.4561 | 2.4597 | 2.4597 | 2.4597 | 2.0763 | 2.0712 | 2.0763 | 2.0712 | 2.0763 | 2.0712 | 3.2304 | 3.1730 | 3.2304 | 3.1730 | 3.2304 | 3.1730 |
N2 | 2.5405 | | 2.4597 | 2.4597 | 2.4597 | 1.4561 | 1.4561 | 1.4561 | 3.2304 | 3.1730 | 3.2304 | 3.1730 | 3.2304 | 3.1730 | 2.0763 | 2.0712 | 2.0763 | 2.0712 | 2.0763 | 2.0712 | C3 | 1.4561 | 2.4597 | | 2.3706 | 2.3706 | 1.5479 | 2.8058 | 2.8547 | 1.0898 | 1.0899 | 3.3133 | 2.5852 | 2.6333 | 3.3110 | 2.1959 | 2.1945 | 3.8160 | 3.1560 | 3.3021 | 3.8225 | C4 | 1.4561 | 2.4597 | 2.3706 | | 2.3706 | 2.8058 | 2.8547 | 1.5479 | 2.6333 | 3.3110 | 1.0898 | 1.0899 | 3.3133 | 2.5852 | 3.8160 | 3.1560 | 3.3021 | 3.8225 | 2.1959 | 2.1945 | C5 | 1.4561 | 2.4597 | 2.3706 | 2.3706 | | 2.8547 | 1.5479 | 2.8058 | 3.3133 | 2.5852 | 2.6333 | 3.3110 | 1.0898 | 1.0899 | 3.3021 | 3.8225 | 2.1959 | 2.1945 | 3.8160 | 3.1560 | C6 | 2.4597 | 1.4561 | 1.5479 | 2.8058 | 2.8547 | | 2.3706 | 2.3706 | 2.1959 | 2.1945 | 3.8160 | 3.1560 | 3.3021 | 3.8225 | 1.0898 | 1.0899 | 3.3133 | 2.5852 | 2.6333 | 3.3110 | C7 | 2.4597 | 1.4561 | 2.8058 | 2.8547 | 1.5479 | 2.3706 | | 2.3706 | 3.8160 | 3.1560 | 3.3021 | 3.8225 | 2.1959 | 2.1945 | 2.6333 | 3.3110 | 1.0898 | 1.0899 | 3.3133 | 2.5852 | C8 | 2.4597 | 1.4561 | 2.8547 | 1.5479 | 2.8058 | 2.3706 | 2.3706 | | 3.3021 | 3.8225 | 2.1959 | 2.1945 | 3.8160 | 3.1560 | 3.3133 | 2.5852 | 2.6333 | 3.3110 | 1.0898 | 1.0899 | H9 | 2.0763 | 3.2304 | 1.0898 | 2.6333 | 3.3133 | 2.1959 | 3.8160 | 3.3021 | | 1.7570 | 3.5945 | 2.3723 | 3.5945 | 4.1275 | 2.9071 | 2.3471 | 4.7735 | 4.2212 | 3.4696 | 4.3442 | H10 | 2.0712 | 3.1730 | 1.0899 | 3.3110 | 2.5852 | 2.1945 | 3.1560 | 3.8225 | 1.7570 | | 4.1275 | 3.5800 | 2.3723 | 3.5800 | 2.3471 | 2.9533 | 4.2212 | 3.1955 | 4.3442 | 4.7136 | H11 | 2.0763 | 3.2304 | 3.3133 | 1.0898 | 2.6333 | 3.8160 | 3.3021 | 2.1959 | 3.5945 | 4.1275 | | 1.7570 | 3.5945 | 2.3723 | 4.7735 | 4.2212 | 3.4696 | 4.3442 | 2.9071 | 2.3471 | H12 | 2.0712 | 3.1730 | 2.5852 | 1.0899 | 3.3110 | 3.1560 | 3.8225 | 2.1945 | 2.3723 | 3.5800 | 1.7570 | | 4.1275 | 3.5800 | 4.2212 | 3.1955 | 4.3442 | 4.7136 | 2.3471 | 2.9533 | H13 | 2.0763 | 3.2304 | 2.6333 | 3.3133 | 1.0898 | 3.3021 | 2.1959 | 3.8160 | 3.5945 | 2.3723 | 3.5945 | 4.1275 | | 1.7570 | 3.4696 | 4.3442 | 2.9071 | 2.3471 | 4.7735 | 4.2212 | H14 | 2.0712 | 3.1730 | 3.3110 | 2.5852 | 1.0899 | 3.8225 | 2.1945 | 3.1560 | 4.1275 | 3.5800 | 2.3723 | 3.5800 | 1.7570 | | 4.3442 | 4.7136 | 2.3471 | 2.9533 | 4.2212 | 3.1955 | H15 | 3.2304 | 2.0763 | 2.1959 | 3.8160 | 3.3021 | 1.0898 | 2.6333 | 3.3133 | 2.9071 | 2.3471 | 4.7735 | 4.2212 | 3.4696 | 4.3442 | | 1.7570 | 3.5945 | 2.3723 | 3.5945 | 4.1275 | H16 | 3.1730 | 2.0712 | 2.1945 | 3.1560 | 3.8225 | 1.0899 | 3.3110 | 2.5852 | 2.3471 | 2.9533 | 4.2212 | 3.1955 | 4.3442 | 4.7136 | 1.7570 | | 4.1275 | 3.5800 | 2.3723 | 3.5800 | H17 | 3.2304 | 2.0763 | 3.8160 | 3.3021 | 2.1959 | 3.3133 | 1.0898 | 2.6333 | 4.7735 | 4.2212 | 3.4696 | 4.3442 | 2.9071 | 2.3471 | 3.5945 | 4.1275 | | 1.7570 | 3.5945 | 2.3723 | H18 | 3.1730 | 2.0712 | 3.1560 | 3.8225 | 2.1945 | 2.5852 | 1.0899 | 3.3110 | 4.2212 | 3.1955 | 4.3442 | 4.7136 | 2.3471 | 2.9533 | 2.3723 | 3.5800 | 1.7570 | | 4.1275 | 3.5800 | H19 | 3.2304 | 2.0763 | 3.3021 | 2.1959 | 3.8160 | 2.6333 | 3.3133 | 1.0898 | 3.4696 | 4.3442 | 2.9071 | 2.3471 | 4.7735 | 4.2212 | 3.5945 | 2.3723 | 3.5945 | 4.1275 | | 1.7570 | H20 | 3.1730 | 2.0712 | 3.8225 | 2.1945 | 3.1560 | 3.3110 | 2.5852 | 1.0899 | 4.3442 | 4.7136 | 2.3471 | 2.9533 | 4.2212 | 3.1955 | 4.1275 | 3.5800 | 2.3723 | 3.5800 | 1.7570 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.891 |
|
N1 |
C3 |
H9 |
108.427 |
| N1 |
C3 |
H10 |
108.024 |
|
N1 |
C4 |
C8 |
109.890 |
| N1 |
C4 |
H11 |
108.427 |
|
N1 |
C4 |
H12 |
108.024 |
| N1 |
C5 |
C7 |
109.891 |
|
N1 |
C5 |
H13 |
108.427 |
| N1 |
C5 |
H14 |
108.024 |
|
N2 |
C6 |
C3 |
109.891 |
| N2 |
C6 |
H15 |
108.427 |
|
N2 |
C6 |
H16 |
108.024 |
| N2 |
C7 |
C5 |
109.890 |
|
N2 |
C7 |
H17 |
108.427 |
| N2 |
C7 |
H18 |
108.024 |
|
N2 |
C8 |
C4 |
109.891 |
| N2 |
C8 |
H19 |
108.427 |
|
N2 |
C8 |
H20 |
108.024 |
| C3 |
N1 |
C4 |
108.987 |
|
C3 |
N1 |
C5 |
108.987 |
| C3 |
C6 |
H15 |
111.526 |
|
C3 |
C6 |
H16 |
111.413 |
| C4 |
N1 |
C5 |
108.987 |
|
C4 |
C8 |
H19 |
111.526 |
| C4 |
C8 |
H20 |
111.413 |
|
C5 |
C6 |
H15 |
104.583 |
| C5 |
C6 |
H16 |
147.960 |
|
C6 |
N2 |
C7 |
108.987 |
| C6 |
N2 |
C8 |
108.987 |
|
C6 |
C3 |
H9 |
111.526 |
| C6 |
C3 |
H10 |
111.413 |
|
C7 |
N2 |
C8 |
108.987 |
| C7 |
C5 |
H13 |
111.526 |
|
C7 |
C5 |
H14 |
111.413 |
| C8 |
C4 |
H11 |
111.526 |
|
C8 |
C4 |
H12 |
111.413 |
| H9 |
C3 |
H10 |
107.433 |
|
H11 |
C4 |
H12 |
107.433 |
| H13 |
C5 |
H14 |
107.433 |
|
H15 |
C6 |
H16 |
107.433 |
| H17 |
C7 |
H18 |
107.433 |
|
H19 |
C8 |
H20 |
107.433 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.148 |
|
|
|
| 2 |
N |
-0.148 |
|
|
|
| 3 |
C |
-0.169 |
|
|
|
| 4 |
C |
-0.169 |
|
|
|
| 5 |
C |
-0.169 |
|
|
|
| 6 |
C |
-0.169 |
|
|
|
| 7 |
C |
-0.169 |
|
|
|
| 8 |
C |
-0.169 |
|
|
|
| 9 |
H |
0.108 |
|
|
|
| 10 |
H |
0.110 |
|
|
|
| 11 |
H |
0.108 |
|
|
|
| 12 |
H |
0.110 |
|
|
|
| 13 |
H |
0.108 |
|
|
|
| 14 |
H |
0.110 |
|
|
|
| 15 |
H |
0.108 |
|
|
|
| 16 |
H |
0.110 |
|
|
|
| 17 |
H |
0.108 |
|
|
|
| 18 |
H |
0.110 |
|
|
|
| 19 |
H |
0.108 |
|
|
|
| 20 |
H |
0.110 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.926 |
0.000 |
0.000 |
| y |
0.000 |
-46.926 |
0.000 |
| z |
0.000 |
0.000 |
-57.169 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.122 |
0.000 |
0.000 |
| y |
0.000 |
5.122 |
0.000 |
| z |
0.000 |
0.000 |
-10.243 |
|
| Polar |
|---|
| 3z2-r2 | -20.486 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.012 |
0.000 |
0.000 |
| y |
0.000 |
12.011 |
0.000 |
| z |
0.000 |
0.000 |
10.925 |
<r2> (average value of r
2) Å
2
| <r2> |
210.956 |
| (<r2>)1/2 |
14.524 |