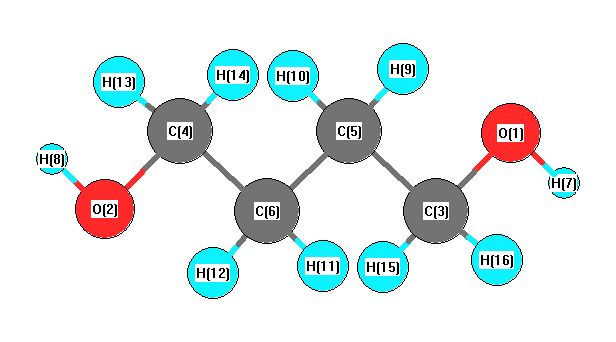
Vibrational Frequencies calculated at B1B95/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3526 |
3367 |
0.00 |
|
|
|
| 2 |
Ag |
3077 |
2938 |
0.00 |
|
|
|
| 3 |
Ag |
2999 |
2864 |
0.00 |
|
|
|
| 4 |
Ag |
1584 |
1513 |
0.00 |
|
|
|
| 5 |
Ag |
1546 |
1476 |
0.00 |
|
|
|
| 6 |
Ag |
1469 |
1402 |
0.00 |
|
|
|
| 7 |
Ag |
1387 |
1325 |
0.00 |
|
|
|
| 8 |
Ag |
1280 |
1222 |
0.00 |
|
|
|
| 9 |
Ag |
1074 |
1025 |
0.00 |
|
|
|
| 10 |
Ag |
1062 |
1014 |
0.00 |
|
|
|
| 11 |
Ag |
1011 |
966 |
0.00 |
|
|
|
| 12 |
Ag |
364 |
348 |
0.00 |
|
|
|
| 13 |
Ag |
315 |
301 |
0.00 |
|
|
|
| 14 |
Au |
3144 |
3002 |
57.14 |
|
|
|
| 15 |
Au |
3029 |
2893 |
115.56 |
|
|
|
| 16 |
Au |
1359 |
1298 |
5.50 |
|
|
|
| 17 |
Au |
1214 |
1159 |
0.83 |
|
|
|
| 18 |
Au |
976 |
932 |
2.26 |
|
|
|
| 19 |
Au |
783 |
748 |
1.50 |
|
|
|
| 20 |
Au |
306 |
292 |
287.57 |
|
|
|
| 21 |
Au |
107 |
102 |
20.08 |
|
|
|
| 22 |
Au |
77 |
74 |
2.23 |
|
|
|
| 23 |
Bg |
3116 |
2976 |
0.00 |
|
|
|
| 24 |
Bg |
3030 |
2893 |
0.00 |
|
|
|
| 25 |
Bg |
1346 |
1285 |
0.00 |
|
|
|
| 26 |
Bg |
1305 |
1246 |
0.00 |
|
|
|
| 27 |
Bg |
1175 |
1122 |
0.00 |
|
|
|
| 28 |
Bg |
848 |
810 |
0.00 |
|
|
|
| 29 |
Bg |
302 |
288 |
0.00 |
|
|
|
| 30 |
Bg |
156 |
149 |
0.00 |
|
|
|
| 31 |
Bu |
3525 |
3366 |
0.45 |
|
|
|
| 32 |
Bu |
3086 |
2947 |
41.42 |
|
|
|
| 33 |
Bu |
2998 |
2863 |
91.37 |
|
|
|
| 34 |
Bu |
1586 |
1514 |
5.69 |
|
|
|
| 35 |
Bu |
1553 |
1483 |
8.08 |
|
|
|
| 36 |
Bu |
1474 |
1408 |
7.83 |
|
|
|
| 37 |
Bu |
1332 |
1272 |
69.67 |
|
|
|
| 38 |
Bu |
1230 |
1175 |
52.27 |
|
|
|
| 39 |
Bu |
1042 |
995 |
154.85 |
|
|
|
| 40 |
Bu |
993 |
948 |
1.24 |
|
|
|
| 41 |
Bu |
503 |
481 |
47.36 |
|
|
|
| 42 |
Bu |
130 |
125 |
8.50 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31209.7 cm
-1
Scaled (by 0.9549) Zero Point Vibrational Energy (zpe) 29802.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.566 |
|
|
|
| 2 |
O |
-0.566 |
|
|
|
| 3 |
C |
-0.158 |
|
|
|
| 4 |
C |
-0.158 |
|
|
|
| 5 |
C |
-0.437 |
|
|
|
| 6 |
C |
-0.437 |
|
|
|
| 7 |
H |
0.345 |
|
|
|
| 8 |
H |
0.345 |
|
|
|
| 9 |
H |
0.222 |
|
|
|
| 10 |
H |
0.222 |
|
|
|
| 11 |
H |
0.222 |
|
|
|
| 12 |
H |
0.222 |
|
|
|
| 13 |
H |
0.186 |
|
|
|
| 14 |
H |
0.186 |
|
|
|
| 15 |
H |
0.186 |
|
|
|
| 16 |
H |
0.186 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.959 |
6.908 |
0.000 |
| y |
6.908 |
-44.164 |
0.000 |
| z |
0.000 |
0.000 |
-37.837 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
16.042 |
6.908 |
0.000 |
| y |
6.908 |
-12.766 |
0.000 |
| z |
0.000 |
0.000 |
-3.276 |
|
| Polar |
|---|
| 3z2-r2 | -6.551 |
|---|
| x2-y2 | 19.205 |
|---|
| xy | 6.908 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.589 |
1.275 |
0.000 |
| y |
1.275 |
7.903 |
0.000 |
| z |
0.000 |
0.000 |
5.805 |
<r2> (average value of r
2) Å
2
| <r2> |
284.394 |
| (<r2>)1/2 |
16.864 |