Jump to
S1C2
Energy calculated at B2PLYP/3-21G
| | hartrees |
|---|
| Energy at 0K | -488.866043 |
| Energy at 298.15K | -488.866366 |
| HF Energy | -488.772966 |
| Nuclear repulsion energy | 78.052361 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3803 |
3803 |
618.04 |
|
|
|
| 2 |
A' |
2159 |
2159 |
431.70 |
|
|
|
| 3 |
A' |
781 |
781 |
18.40 |
|
|
|
| 4 |
A' |
511 |
511 |
0.91 |
|
|
|
| 5 |
A' |
175 |
175 |
293.70 |
|
|
|
| 6 |
A" |
514 |
514 |
0.16 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3971.1 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 3971.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.031 |
1.713 |
0.000 |
| C2 |
0.000 |
0.528 |
0.000 |
| S3 |
-0.001 |
-1.115 |
0.000 |
| H4 |
0.236 |
2.673 |
0.000 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
S3 |
H4 |
| N1 | | 1.1856 | 2.8279 | 0.9966 |
C2 | 1.1856 | | 1.6425 | 2.1584 | S3 | 2.8279 | 1.6425 | | 3.7953 | H4 | 0.9966 | 2.1584 | 3.7953 | |
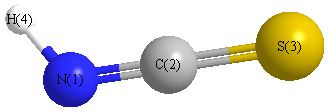 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
S3 |
178.456 |
|
C2 |
N1 |
H4 |
162.956 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.734 |
|
|
|
| 2 |
C |
0.148 |
|
|
|
| 3 |
S |
0.201 |
|
|
|
| 4 |
H |
0.385 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.591 |
3.945 |
0.000 |
3.989 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.267 |
1.588 |
0.000 |
| y |
1.588 |
-16.485 |
0.000 |
| z |
0.000 |
0.000 |
-25.426 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.312 |
1.588 |
0.000 |
| y |
1.588 |
8.861 |
0.000 |
| z |
0.000 |
0.000 |
-4.549 |
|
| Polar |
|---|
| 3z2-r2 | -9.099 |
|---|
| x2-y2 | -8.782 |
|---|
| xy | 1.588 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.691 |
-0.039 |
0.000 |
| y |
-0.039 |
8.339 |
0.000 |
| z |
0.000 |
0.000 |
1.661 |
<r2> (average value of r
2) Å
2
| <r2> |
63.287 |
| (<r2>)1/2 |
7.955 |
Jump to
S1C1
Energy calculated at B2PLYP/3-21G
| | hartrees |
|---|
| Energy at 0K | -488.866009 |
| Energy at 298.15K | |
| HF Energy | -488.773594 |
| Nuclear repulsion energy | 78.036778 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3832 |
3832 |
708.20 |
|
|
|
| 2 |
Σ |
2173 |
2173 |
424.35 |
|
|
|
| 3 |
Σ |
776 |
776 |
24.46 |
|
|
|
| 4 |
Π |
517 |
517 |
0.20 |
|
|
|
| 4 |
Π |
517 |
517 |
0.20 |
|
|
|
| 5 |
Π |
116i |
116i |
224.70 |
|
|
|
| 5 |
Π |
116i |
116i |
224.70 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3791.3 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 3791.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/3-21G
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
-1.711 |
| C2 |
0.000 |
0.000 |
-0.529 |
| S3 |
0.000 |
0.000 |
1.116 |
| H4 |
0.000 |
0.000 |
-2.706 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
S3 |
H4 |
| N1 | | 1.1828 | 2.8276 | 0.9941 |
C2 | 1.1828 | | 1.6448 | 2.1769 | S3 | 2.8276 | 1.6448 | | 3.8217 | H4 | 0.9941 | 2.1769 | 3.8217 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
S3 |
180.000 |
|
C2 |
N1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.749 |
|
|
|
| 2 |
C |
0.167 |
|
|
|
| 3 |
S |
0.192 |
|
|
|
| 4 |
H |
0.390 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.157 |
4.157 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.422 |
0.000 |
0.000 |
| y |
0.000 |
-25.422 |
0.000 |
| z |
0.000 |
0.000 |
-15.911 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.756 |
0.000 |
0.000 |
| y |
0.000 |
-4.756 |
0.000 |
| z |
0.000 |
0.000 |
9.511 |
|
| Polar |
|---|
| 3z2-r2 | 19.022 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.661 |
0.000 |
0.000 |
| y |
0.000 |
1.661 |
0.000 |
| z |
0.000 |
0.000 |
8.257 |
<r2> (average value of r
2) Å
2
| <r2> |
63.332 |
| (<r2>)1/2 |
7.958 |