Jump to
S1C2
S1C3
S1C4
Energy calculated at B2PLYP/STO-3G
| | hartrees |
|---|
| Energy at 0K | -572.958761 |
| Energy at 298.15K | |
| HF Energy | -572.910410 |
| Nuclear repulsion energy | 70.453184 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2745 |
2745 |
0.00 |
225.01 |
0.29 |
0.45 |
| 2 |
Σg |
918 |
918 |
0.00 |
19.58 |
0.26 |
0.41 |
| 3 |
Σu |
2738 |
2738 |
16.09 |
0.00 |
0.00 |
0.00 |
| 4 |
Πg |
744i |
744i |
0.00 |
2.98 |
0.75 |
0.86 |
| 4 |
Πg |
744i |
744i |
0.00 |
2.98 |
0.75 |
0.86 |
| 5 |
Πu |
483 |
483 |
13.93 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
483 |
483 |
13.93 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2939.1 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 2939.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/STO-3G
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
0.935 |
| Si2 |
0.000 |
0.000 |
-0.935 |
| H3 |
0.000 |
0.000 |
2.361 |
| H4 |
0.000 |
0.000 |
-2.361 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 1.8705 | 1.4253 | 3.2958 |
Si2 | 1.8705 | | 3.2958 | 1.4253 | H3 | 1.4253 | 3.2958 | | 4.7211 | H4 | 3.2958 | 1.4253 | 4.7211 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.080 |
|
|
|
| 2 |
Si |
0.080 |
|
|
|
| 3 |
H |
-0.080 |
|
|
|
| 4 |
H |
-0.080 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-22.242 |
0.000 |
0.000 |
| y |
0.000 |
-22.242 |
0.000 |
| z |
0.000 |
0.000 |
-21.623 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.309 |
0.000 |
0.000 |
| y |
0.000 |
-0.309 |
0.000 |
| z |
0.000 |
0.000 |
0.618 |
|
| Polar |
|---|
| 3z2-r2 | 1.236 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.802 |
0.000 |
0.000 |
| y |
0.000 |
0.802 |
0.000 |
| z |
0.000 |
0.000 |
6.821 |
<r2> (average value of r
2) Å
2
| <r2> |
49.399 |
| (<r2>)1/2 |
7.028 |
Jump to
S1C1
S1C3
S1C4
Energy calculated at B2PLYP/STO-3G
| | hartrees |
|---|
| Energy at 0K | -573.013494 |
| Energy at 298.15K | |
| HF Energy | -572.965212 |
| Nuclear repulsion energy | 66.864432 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2612 |
2612 |
0.00 |
238.98 |
0.29 |
0.45 |
| 2 |
Ag |
770 |
770 |
0.00 |
13.14 |
0.71 |
0.83 |
| 3 |
Ag |
714 |
714 |
0.00 |
14.99 |
0.47 |
0.64 |
| 4 |
Au |
558 |
558 |
2.48 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2619 |
2619 |
66.98 |
0.00 |
0.00 |
0.00 |
| 6 |
Bu |
440 |
440 |
11.00 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3856.4 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 3856.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/STO-3G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.003 |
0.000 |
| Si2 |
0.000 |
-1.003 |
0.000 |
| H3 |
1.227 |
1.786 |
0.000 |
| H4 |
-1.227 |
-1.786 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.0060 | 1.4559 | 3.0473 |
Si2 | 2.0060 | | 3.0473 | 1.4559 | H3 | 1.4559 | 3.0473 | | 4.3345 | H4 | 3.0473 | 1.4559 | 4.3345 | |
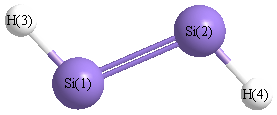 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
122.544 |
|
Si2 |
Si1 |
H3 |
122.544 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.136 |
|
|
|
| 2 |
Si |
0.136 |
|
|
|
| 3 |
H |
-0.136 |
|
|
|
| 4 |
H |
-0.136 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.322 |
-1.624 |
0.000 |
| y |
-1.624 |
-21.388 |
0.000 |
| z |
0.000 |
0.000 |
-22.724 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.266 |
-1.624 |
0.000 |
| y |
-1.624 |
1.635 |
0.000 |
| z |
0.000 |
0.000 |
-0.369 |
|
| Polar |
|---|
| 3z2-r2 | -0.739 |
|---|
| x2-y2 | -1.934 |
|---|
| xy | -1.624 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.407 |
0.354 |
0.000 |
| y |
0.354 |
7.215 |
0.000 |
| z |
0.000 |
0.000 |
1.422 |
<r2> (average value of r
2) Å
2
| <r2> |
51.603 |
| (<r2>)1/2 |
7.183 |
Jump to
S1C1
S1C2
S1C4
Energy calculated at B2PLYP/STO-3G
| | hartrees |
|---|
| Energy at 0K | -573.026727 |
| Energy at 298.15K | |
| HF Energy | -572.995530 |
| Nuclear repulsion energy | 66.423210 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1949 |
1949 |
6.90 |
41.43 |
0.14 |
0.25 |
| 2 |
A1 |
1326 |
1326 |
54.18 |
11.99 |
0.52 |
0.69 |
| 3 |
A1 |
699 |
699 |
0.00 |
21.56 |
0.36 |
0.53 |
| 4 |
A2 |
920 |
920 |
0.00 |
3.11 |
0.75 |
0.86 |
| 5 |
B1 |
1911 |
1911 |
39.76 |
34.16 |
0.75 |
0.86 |
| 6 |
B2 |
1085 |
1085 |
559.58 |
5.85 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3944.3 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 3944.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/STO-3G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.070 |
-0.056 |
| Si2 |
0.000 |
-1.070 |
-0.056 |
| H3 |
0.976 |
0.000 |
0.784 |
| H4 |
-0.976 |
0.000 |
0.784 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1407 | 1.6742 | 1.6742 |
Si2 | 2.1407 | | 1.6742 | 1.6742 | H3 | 1.6742 | 1.6742 | | 1.9516 | H4 | 1.6742 | 1.6742 | 1.9516 | |
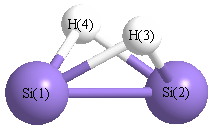 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
50.259 |
|
Si2 |
Si1 |
H3 |
50.259 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.177 |
|
|
|
| 2 |
Si |
0.177 |
|
|
|
| 3 |
H |
-0.177 |
|
|
|
| 4 |
H |
-0.177 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.516 |
0.516 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-21.824 |
0.000 |
0.000 |
| y |
0.000 |
-26.134 |
0.000 |
| z |
0.000 |
0.000 |
-22.662 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.574 |
0.000 |
0.000 |
| y |
0.000 |
-3.891 |
0.000 |
| z |
0.000 |
0.000 |
1.317 |
|
| Polar |
|---|
| 3z2-r2 | 2.634 |
|---|
| x2-y2 | 4.310 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.762 |
0.000 |
0.000 |
| y |
0.000 |
5.029 |
0.000 |
| z |
0.000 |
0.000 |
2.343 |
<r2> (average value of r
2) Å
2
| <r2> |
50.000 |
| (<r2>)1/2 |
7.071 |
Jump to
S1C1
S1C2
S1C3
Energy calculated at B2PLYP/STO-3G
| | hartrees |
|---|
| Energy at 0K | -573.011046 |
| Energy at 298.15K | |
| HF Energy | -572.971081 |
| Nuclear repulsion energy | 67.556282 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2616 |
2616 |
35.36 |
172.64 |
0.32 |
0.49 |
| 2 |
A' |
2077 |
2077 |
55.34 |
48.83 |
0.26 |
0.41 |
| 3 |
A' |
992 |
992 |
151.82 |
11.32 |
0.74 |
0.85 |
| 4 |
A' |
749 |
749 |
30.98 |
16.51 |
0.27 |
0.42 |
| 5 |
A' |
475 |
475 |
27.55 |
0.36 |
0.27 |
0.42 |
| 6 |
A" |
92 |
92 |
15.26 |
1.53 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3500.0 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 3500.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/STO-3G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.057 |
-1.098 |
0.000 |
| Si2 |
0.057 |
0.926 |
0.000 |
| H3 |
-1.280 |
0.082 |
0.000 |
| H4 |
-0.310 |
2.329 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.0241 | 1.7833 | 3.4472 |
Si2 | 2.0241 | | 1.5804 | 1.4507 | H3 | 1.7833 | 1.5804 | | 2.4474 | H4 | 3.4472 | 1.4507 | 2.4474 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
165.358 |
|
Si2 |
Si1 |
H3 |
48.542 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.154 |
|
|
|
| 2 |
Si |
0.123 |
|
|
|
| 3 |
H |
-0.157 |
|
|
|
| 4 |
H |
-0.120 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.244 |
0.190 |
0.000 |
0.309 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-21.934 |
0.195 |
-0.000 |
| y |
0.195 |
-23.699 |
0.003 |
| z |
-0.000 |
0.003 |
-22.976 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.403 |
0.195 |
-0.000 |
| y |
0.195 |
-1.244 |
0.003 |
| z |
-0.000 |
0.003 |
-0.159 |
|
| Polar |
|---|
| 3z2-r2 | -0.319 |
|---|
| x2-y2 | 1.765 |
|---|
| xy | 0.195 |
|---|
| xz | -0.000 |
|---|
| yz | 0.003 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.560 |
-0.075 |
0.000 |
| y |
-0.075 |
6.670 |
0.002 |
| z |
0.000 |
0.002 |
1.725 |
<r2> (average value of r
2) Å
2
| <r2> |
50.428 |
| (<r2>)1/2 |
7.101 |