Jump to
S1C2
S1C3
Energy calculated at B2PLYP/3-21G*
| | hartrees |
|---|
| Energy at 0K | -99.682374 |
| Energy at 298.15K | -99.681552 |
| HF Energy | -99.607199 |
| Nuclear repulsion energy | 27.327821 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at B2PLYP/3-21G*
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.075 |
| C2 |
0.000 |
0.000 |
-0.159 |
| N3 |
0.000 |
0.000 |
1.025 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9165 | 3.1004 |
C2 | 1.9165 | | 1.1839 | N3 | 3.1004 | 1.1839 | |
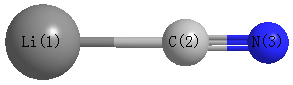 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Li |
0.503 |
|
|
|
| 2 |
C |
-0.070 |
|
|
|
| 3 |
N |
-0.433 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-8.486 |
8.486 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-14.438 |
0.000 |
0.000 |
| y |
0.000 |
-14.438 |
0.000 |
| z |
0.000 |
0.000 |
0.873 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.655 |
0.000 |
0.000 |
| y |
0.000 |
-7.655 |
0.000 |
| z |
0.000 |
0.000 |
15.311 |
|
| Polar |
|---|
| 3z2-r2 | 30.621 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.151 |
0.000 |
0.000 |
| y |
0.000 |
2.151 |
0.000 |
| z |
0.000 |
0.000 |
4.065 |
<r2> (average value of r
2) Å
2
| <r2> |
26.258 |
| (<r2>)1/2 |
5.124 |
Jump to
S1C1
S1C3
Energy calculated at B2PLYP/3-21G*
| | hartrees |
|---|
| Energy at 0K | -99.690269 |
| Energy at 298.15K | |
| HF Energy | -99.620056 |
| Nuclear repulsion energy | 28.234659 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/3-21G*
Geometric Data calculated at B2PLYP/3-21G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.470 |
-0.541 |
0.000 |
| C2 |
-0.735 |
-0.384 |
0.000 |
| N3 |
0.000 |
0.561 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.2100 | 1.8367 |
C2 | 2.2100 | | 1.1966 | N3 | 1.8367 | 1.1966 | |
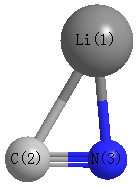 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
56.198 |
|
Li1 |
N3 |
C2 |
91.027 |
| C2 |
Li1 |
N3 |
32.775 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Li |
0.505 |
|
|
|
| 2 |
C |
0.089 |
|
|
|
| 3 |
N |
-0.594 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.308 |
7.576 |
0.000 |
8.267 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-12.304 |
4.360 |
0.000 |
| y |
4.360 |
-5.255 |
0.000 |
| z |
0.000 |
0.000 |
-14.334 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.509 |
4.360 |
0.000 |
| y |
4.360 |
8.064 |
0.000 |
| z |
0.000 |
0.000 |
-5.554 |
|
| Polar |
|---|
| 3z2-r2 | -11.109 |
|---|
| x2-y2 | -7.049 |
|---|
| xy | 4.360 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.151 |
0.000 |
0.000 |
| y |
0.000 |
2.151 |
0.000 |
| z |
0.000 |
0.000 |
4.065 |
<r2> (average value of r
2) Å
2
| <r2> |
24.020 |
| (<r2>)1/2 |
4.901 |
Jump to
S1C1
S1C2
Energy calculated at B2PLYP/3-21G*
| | hartrees |
|---|
| Energy at 0K | -99.690398 |
| Energy at 298.15K | -99.689368 |
| HF Energy | -99.620114 |
| Nuclear repulsion energy | 28.150630 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at B2PLYP/3-21G*
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.876 |
| C2 |
0.000 |
0.000 |
-1.076 |
| N3 |
0.000 |
0.000 |
0.118 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9526 | 1.7578 |
C2 | 2.9526 | | 1.1947 | N3 | 1.7578 | 1.1947 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Li |
0.506 |
|
|
|
| 2 |
C |
0.086 |
|
|
|
| 3 |
N |
-0.592 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
8.358 |
8.358 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-14.338 |
0.000 |
0.000 |
| y |
0.000 |
-14.338 |
0.000 |
| z |
0.000 |
0.000 |
-2.982 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.678 |
0.000 |
0.000 |
| y |
0.000 |
-5.678 |
0.000 |
| z |
0.000 |
0.000 |
11.356 |
|
| Polar |
|---|
| 3z2-r2 | 22.712 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.029 |
0.000 |
0.000 |
| y |
0.000 |
2.029 |
0.000 |
| z |
0.000 |
0.000 |
4.443 |
<r2> (average value of r
2) Å
2
| <r2> |
24.201 |
| (<r2>)1/2 |
4.919 |