Jump to
S1C2
Vibrational Frequencies calculated at B2PLYP/6-31G
Geometric Data calculated at B2PLYP/6-31G
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B2PLYP/6-31G
| | hartrees |
|---|
| Energy at 0K | -344.681378 |
| Energy at 298.15K | -344.698482 |
| HF Energy | -344.424732 |
| Nuclear repulsion energy | 419.991246 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3112 |
3112 |
0.00 |
|
|
|
| 2 |
A1 |
3083 |
3083 |
0.00 |
|
|
|
| 3 |
A1 |
1552 |
1552 |
0.00 |
|
|
|
| 4 |
A1 |
1369 |
1369 |
0.00 |
|
|
|
| 5 |
A1 |
1294 |
1294 |
0.00 |
|
|
|
| 6 |
A1 |
1039 |
1039 |
0.00 |
|
|
|
| 7 |
A1 |
951 |
951 |
0.00 |
|
|
|
| 8 |
A1 |
791 |
791 |
0.00 |
|
|
|
| 9 |
A1 |
609 |
609 |
0.00 |
|
|
|
| 10 |
A1 |
93 |
93 |
0.00 |
|
|
|
| 11 |
A2 |
3134 |
3134 |
0.00 |
|
|
|
| 12 |
A2 |
3069 |
3069 |
113.56 |
|
|
|
| 13 |
A2 |
1547 |
1547 |
5.31 |
|
|
|
| 14 |
A2 |
1407 |
1407 |
1.51 |
|
|
|
| 15 |
A2 |
1212 |
1212 |
0.00 |
|
|
|
| 16 |
A2 |
995 |
995 |
20.76 |
|
|
|
| 17 |
A2 |
829 |
829 |
0.00 |
|
|
|
| 18 |
A2 |
707 |
707 |
68.89 |
|
|
|
| 19 |
E |
3143 |
3143 |
84.25 |
|
|
|
| 19 |
E |
3143 |
3143 |
84.32 |
|
|
|
| 20 |
E |
3117 |
3117 |
0.01 |
|
|
|
| 20 |
E |
3117 |
3117 |
0.01 |
|
|
|
| 21 |
E |
3076 |
3076 |
98.32 |
|
|
|
| 21 |
E |
3076 |
3076 |
98.21 |
|
|
|
| 22 |
E |
3066 |
3066 |
0.00 |
|
|
|
| 22 |
E |
3066 |
3066 |
0.00 |
|
|
|
| 23 |
E |
1544 |
1544 |
12.45 |
|
|
|
| 23 |
E |
1544 |
1544 |
12.46 |
|
|
|
| 24 |
E |
1536 |
1536 |
0.00 |
|
|
|
| 24 |
E |
1536 |
1536 |
0.00 |
|
|
|
| 25 |
E |
1375 |
1375 |
0.00 |
|
|
|
| 25 |
E |
1375 |
1375 |
0.00 |
|
|
|
| 26 |
E |
1372 |
1372 |
5.72 |
|
|
|
| 26 |
E |
1372 |
1372 |
5.72 |
|
|
|
| 27 |
E |
1351 |
1351 |
0.00 |
|
|
|
| 27 |
E |
1351 |
1351 |
0.00 |
|
|
|
| 28 |
E |
1347 |
1347 |
0.10 |
|
|
|
| 28 |
E |
1347 |
1347 |
0.10 |
|
|
|
| 29 |
E |
1228 |
1228 |
0.00 |
|
|
|
| 29 |
E |
1228 |
1228 |
0.00 |
|
|
|
| 30 |
E |
1060 |
1060 |
31.35 |
|
|
|
| 30 |
E |
1060 |
1060 |
31.35 |
|
|
|
| 31 |
E |
1023 |
1023 |
0.00 |
|
|
|
| 31 |
E |
1023 |
1023 |
0.00 |
|
|
|
| 32 |
E |
899 |
899 |
3.31 |
|
|
|
| 32 |
E |
899 |
899 |
3.31 |
|
|
|
| 33 |
E |
847 |
847 |
7.18 |
|
|
|
| 33 |
E |
847 |
847 |
7.18 |
|
|
|
| 34 |
E |
584 |
584 |
0.00 |
|
|
|
| 34 |
E |
584 |
584 |
0.00 |
|
|
|
| 35 |
E |
426 |
426 |
0.04 |
|
|
|
| 35 |
E |
426 |
426 |
0.04 |
|
|
|
| 36 |
E |
327 |
327 |
0.00 |
|
|
|
| 36 |
E |
327 |
327 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40714.6 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 40714.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/6-31G
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.286 |
| N2 |
0.000 |
0.000 |
-1.286 |
| C3 |
-0.001 |
1.408 |
0.785 |
| C4 |
1.220 |
-0.703 |
0.785 |
| C5 |
-1.218 |
-0.705 |
0.785 |
| C6 |
0.001 |
1.408 |
-0.785 |
| C7 |
-1.220 |
-0.703 |
-0.785 |
| C8 |
1.218 |
-0.705 |
-0.785 |
| H9 |
0.882 |
1.910 |
1.193 |
| H10 |
-0.888 |
1.906 |
1.190 |
| H11 |
1.213 |
-1.719 |
1.193 |
| H12 |
2.095 |
-0.184 |
1.190 |
| H13 |
-2.095 |
-0.191 |
1.193 |
| H14 |
-1.206 |
-1.722 |
1.190 |
| H15 |
-0.882 |
1.910 |
-1.193 |
| H16 |
0.888 |
1.906 |
-1.190 |
| H17 |
-1.213 |
-1.719 |
-1.193 |
| H18 |
-2.095 |
-0.184 |
-1.190 |
| H19 |
2.095 |
-0.191 |
-1.193 |
| H20 |
1.206 |
-1.722 |
-1.190 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5720 | 1.4940 | 1.4940 | 1.4940 | 2.5044 | 2.5044 | 2.5044 | 2.1055 | 2.1052 | 2.1055 | 2.1052 | 2.1055 | 2.1052 | 3.2514 | 3.2485 | 3.2514 | 3.2485 | 3.2514 | 3.2485 |
N2 | 2.5720 | | 2.5044 | 2.5044 | 2.5044 | 1.4940 | 1.4940 | 1.4940 | 3.2514 | 3.2485 | 3.2514 | 3.2485 | 3.2514 | 3.2485 | 2.1055 | 2.1052 | 2.1055 | 2.1052 | 2.1055 | 2.1052 | C3 | 1.4940 | 2.5044 | | 2.4381 | 2.4381 | 1.5706 | 2.8990 | 2.9014 | 1.0950 | 1.0950 | 3.3786 | 2.6628 | 2.6654 | 3.3784 | 2.2231 | 2.2230 | 3.8931 | 3.2888 | 3.2961 | 3.8933 | C4 | 1.4940 | 2.5044 | 2.4381 | | 2.4381 | 2.8990 | 2.9014 | 1.5706 | 2.6654 | 3.3784 | 1.0950 | 1.0950 | 3.3786 | 2.6628 | 3.8931 | 3.2888 | 3.2961 | 3.8933 | 2.2231 | 2.2230 | C5 | 1.4940 | 2.5044 | 2.4381 | 2.4381 | | 2.9014 | 1.5706 | 2.8990 | 3.3786 | 2.6628 | 2.6654 | 3.3784 | 1.0950 | 1.0950 | 3.2961 | 3.8933 | 2.2231 | 2.2230 | 3.8931 | 3.2888 | C6 | 2.5044 | 1.4940 | 1.5706 | 2.8990 | 2.9014 | | 2.4381 | 2.4381 | 2.2231 | 2.2230 | 3.8931 | 3.2888 | 3.2961 | 3.8933 | 1.0950 | 1.0950 | 3.3786 | 2.6628 | 2.6654 | 3.3784 | C7 | 2.5044 | 1.4940 | 2.8990 | 2.9014 | 1.5706 | 2.4381 | | 2.4381 | 3.8931 | 3.2888 | 3.2961 | 3.8933 | 2.2231 | 2.2230 | 2.6654 | 3.3784 | 1.0950 | 1.0950 | 3.3786 | 2.6628 | C8 | 2.5044 | 1.4940 | 2.9014 | 1.5706 | 2.8990 | 2.4381 | 2.4381 | | 3.2961 | 3.8933 | 2.2231 | 2.2230 | 3.8931 | 3.2888 | 3.3786 | 2.6628 | 2.6654 | 3.3784 | 1.0950 | 1.0950 | H9 | 2.1055 | 3.2514 | 1.0950 | 2.6654 | 3.3786 | 2.2231 | 3.8931 | 3.2961 | | 1.7706 | 3.6434 | 2.4192 | 3.6434 | 4.1898 | 2.9678 | 2.3831 | 4.8217 | 4.3501 | 3.4027 | 4.3562 | H10 | 2.1052 | 3.2485 | 1.0950 | 3.3784 | 2.6628 | 2.2230 | 3.2888 | 3.8933 | 1.7706 | | 4.1898 | 3.6425 | 2.4192 | 3.6425 | 2.3831 | 2.9700 | 4.3501 | 3.3891 | 4.3562 | 4.8186 | H11 | 2.1055 | 3.2514 | 3.3786 | 1.0950 | 2.6654 | 3.8931 | 3.2961 | 2.2231 | 3.6434 | 4.1898 | | 1.7706 | 3.6434 | 2.4192 | 4.8217 | 4.3501 | 3.4027 | 4.3562 | 2.9678 | 2.3831 | H12 | 2.1052 | 3.2485 | 2.6628 | 1.0950 | 3.3784 | 3.2888 | 3.8933 | 2.2230 | 2.4192 | 3.6425 | 1.7706 | | 4.1898 | 3.6425 | 4.3501 | 3.3891 | 4.3562 | 4.8186 | 2.3831 | 2.9700 | H13 | 2.1055 | 3.2514 | 2.6654 | 3.3786 | 1.0950 | 3.2961 | 2.2231 | 3.8931 | 3.6434 | 2.4192 | 3.6434 | 4.1898 | | 1.7706 | 3.4027 | 4.3562 | 2.9678 | 2.3831 | 4.8217 | 4.3501 | H14 | 2.1052 | 3.2485 | 3.3784 | 2.6628 | 1.0950 | 3.8933 | 2.2230 | 3.2888 | 4.1898 | 3.6425 | 2.4192 | 3.6425 | 1.7706 | | 4.3562 | 4.8186 | 2.3831 | 2.9700 | 4.3501 | 3.3891 | H15 | 3.2514 | 2.1055 | 2.2231 | 3.8931 | 3.2961 | 1.0950 | 2.6654 | 3.3786 | 2.9678 | 2.3831 | 4.8217 | 4.3501 | 3.4027 | 4.3562 | | 1.7706 | 3.6434 | 2.4192 | 3.6434 | 4.1898 | H16 | 3.2485 | 2.1052 | 2.2230 | 3.2888 | 3.8933 | 1.0950 | 3.3784 | 2.6628 | 2.3831 | 2.9700 | 4.3501 | 3.3891 | 4.3562 | 4.8186 | 1.7706 | | 4.1898 | 3.6425 | 2.4192 | 3.6425 | H17 | 3.2514 | 2.1055 | 3.8931 | 3.2961 | 2.2231 | 3.3786 | 1.0950 | 2.6654 | 4.8217 | 4.3501 | 3.4027 | 4.3562 | 2.9678 | 2.3831 | 3.6434 | 4.1898 | | 1.7706 | 3.6434 | 2.4192 | H18 | 3.2485 | 2.1052 | 3.2888 | 3.8933 | 2.2230 | 2.6628 | 1.0950 | 3.3784 | 4.3501 | 3.3891 | 4.3562 | 4.8186 | 2.3831 | 2.9700 | 2.4192 | 3.6425 | 1.7706 | | 4.1898 | 3.6425 | H19 | 3.2514 | 2.1055 | 3.2961 | 2.2231 | 3.8931 | 2.6654 | 3.3786 | 1.0950 | 3.4027 | 4.3562 | 2.9678 | 2.3831 | 4.8217 | 4.3501 | 3.6434 | 2.4192 | 3.6434 | 4.1898 | | 1.7706 | H20 | 3.2485 | 2.1052 | 3.8933 | 2.2230 | 3.2888 | 3.3784 | 2.6628 | 1.0950 | 4.3562 | 4.8186 | 2.3831 | 2.9700 | 4.3501 | 3.3891 | 4.1898 | 3.6425 | 2.4192 | 3.6425 | 1.7706 | |
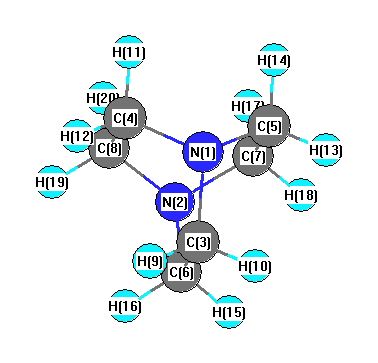 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.581 |
|
N1 |
C3 |
H9 |
107.837 |
| N1 |
C3 |
H10 |
107.808 |
|
N1 |
C4 |
C8 |
109.581 |
| N1 |
C4 |
H11 |
107.837 |
|
N1 |
C4 |
H12 |
107.808 |
| N1 |
C5 |
C7 |
109.581 |
|
N1 |
C5 |
H13 |
107.837 |
| N1 |
C5 |
H14 |
107.808 |
|
N2 |
C6 |
C3 |
109.581 |
| N2 |
C6 |
H15 |
107.837 |
|
N2 |
C6 |
H16 |
107.808 |
| N2 |
C7 |
C5 |
109.581 |
|
N2 |
C7 |
H17 |
107.837 |
| N2 |
C7 |
H18 |
107.808 |
|
N2 |
C8 |
C4 |
109.581 |
| N2 |
C8 |
H19 |
107.837 |
|
N2 |
C8 |
H20 |
107.808 |
| C3 |
N1 |
C4 |
109.361 |
|
C3 |
N1 |
C5 |
109.361 |
| C3 |
C6 |
H15 |
111.784 |
|
C3 |
C6 |
H16 |
111.773 |
| C4 |
N1 |
C5 |
109.361 |
|
C4 |
C8 |
H19 |
111.784 |
| C4 |
C8 |
H20 |
111.773 |
|
C5 |
C6 |
H15 |
101.320 |
| C5 |
C6 |
H16 |
150.697 |
|
C6 |
N2 |
C7 |
109.361 |
| C6 |
N2 |
C8 |
109.361 |
|
C6 |
C3 |
H9 |
111.784 |
| C6 |
C3 |
H10 |
111.773 |
|
C7 |
N2 |
C8 |
109.361 |
| C7 |
C5 |
H13 |
111.784 |
|
C7 |
C5 |
H14 |
111.773 |
| C8 |
C4 |
H11 |
111.784 |
|
C8 |
C4 |
H12 |
111.773 |
| H9 |
C3 |
H10 |
107.897 |
|
H11 |
C4 |
H12 |
107.897 |
| H13 |
C5 |
H14 |
107.897 |
|
H15 |
C6 |
H16 |
107.897 |
| H17 |
C7 |
H18 |
107.897 |
|
H19 |
C8 |
H20 |
107.897 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.431 |
|
|
|
| 2 |
N |
-0.431 |
|
|
|
| 3 |
C |
-0.149 |
|
|
|
| 4 |
C |
-0.149 |
|
|
|
| 5 |
C |
-0.149 |
|
|
|
| 6 |
C |
-0.149 |
|
|
|
| 7 |
C |
-0.149 |
|
|
|
| 8 |
C |
-0.149 |
|
|
|
| 9 |
H |
0.146 |
|
|
|
| 10 |
H |
0.146 |
|
|
|
| 11 |
H |
0.146 |
|
|
|
| 12 |
H |
0.146 |
|
|
|
| 13 |
H |
0.146 |
|
|
|
| 14 |
H |
0.146 |
|
|
|
| 15 |
H |
0.146 |
|
|
|
| 16 |
H |
0.146 |
|
|
|
| 17 |
H |
0.146 |
|
|
|
| 18 |
H |
0.146 |
|
|
|
| 19 |
H |
0.146 |
|
|
|
| 20 |
H |
0.146 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.850 |
0.000 |
0.000 |
| y |
0.000 |
-46.850 |
0.000 |
| z |
0.000 |
0.000 |
-59.849 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.500 |
0.000 |
0.000 |
| y |
0.000 |
6.500 |
0.000 |
| z |
0.000 |
0.000 |
-12.999 |
|
| Polar |
|---|
| 3z2-r2 | -25.998 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.860 |
0.000 |
0.000 |
| y |
0.000 |
10.861 |
0.000 |
| z |
0.000 |
0.000 |
9.140 |
<r2> (average value of r
2) Å
2
| <r2> |
218.777 |
| (<r2>)1/2 |
14.791 |