Vibrational Frequencies calculated at HSEh1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3689 |
3509 |
90.79 |
|
|
|
| 2 |
A' |
3287 |
3126 |
1.14 |
|
|
|
| 3 |
A' |
3224 |
3066 |
19.07 |
|
|
|
| 4 |
A' |
3212 |
3055 |
16.06 |
|
|
|
| 5 |
A' |
1696 |
1613 |
66.67 |
|
|
|
| 6 |
A' |
1655 |
1574 |
80.49 |
|
|
|
| 7 |
A' |
1565 |
1488 |
29.56 |
|
|
|
| 8 |
A' |
1517 |
1443 |
3.93 |
|
|
|
| 9 |
A' |
1462 |
1391 |
65.46 |
|
|
|
| 10 |
A' |
1445 |
1374 |
20.23 |
|
|
|
| 11 |
A' |
1395 |
1327 |
65.73 |
|
|
|
| 12 |
A' |
1345 |
1279 |
16.22 |
|
|
|
| 13 |
A' |
1338 |
1272 |
14.13 |
|
|
|
| 14 |
A' |
1305 |
1241 |
30.74 |
|
|
|
| 15 |
A' |
1224 |
1164 |
7.18 |
|
|
|
| 16 |
A' |
1157 |
1100 |
6.14 |
|
|
|
| 17 |
A' |
1108 |
1054 |
13.32 |
|
|
|
| 18 |
A' |
954 |
907 |
1.43 |
|
|
|
| 19 |
A' |
920 |
875 |
12.15 |
|
|
|
| 20 |
A' |
819 |
779 |
14.71 |
|
|
|
| 21 |
A' |
664 |
631 |
0.45 |
|
|
|
| 22 |
A' |
573 |
545 |
4.04 |
|
|
|
| 23 |
A' |
448 |
426 |
12.80 |
|
|
|
| 24 |
A" |
999 |
950 |
0.44 |
|
|
|
| 25 |
A" |
946 |
900 |
9.04 |
|
|
|
| 26 |
A" |
885 |
842 |
5.51 |
|
|
|
| 27 |
A" |
815 |
775 |
9.40 |
|
|
|
| 28 |
A" |
672 |
639 |
5.97 |
|
|
|
| 29 |
A" |
628 |
597 |
26.28 |
|
|
|
| 30 |
A" |
540 |
514 |
114.57 |
|
|
|
| 31 |
A" |
420 |
400 |
3.27 |
|
|
|
| 32 |
A" |
249 |
237 |
0.29 |
|
|
|
| 33 |
A" |
232 |
221 |
4.45 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21193.3 cm
-1
Scaled (by 0.951) Zero Point Vibrational Energy (zpe) 20154.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
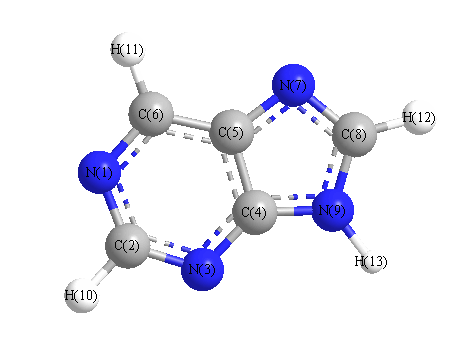
Charges from optimized geometry at HSEh1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.424 |
|
|
|
| 2 |
C |
0.145 |
|
|
|
| 3 |
N |
-0.484 |
|
|
|
| 4 |
C |
0.539 |
|
|
|
| 5 |
C |
0.188 |
|
|
|
| 6 |
C |
0.029 |
|
|
|
| 7 |
N |
-0.496 |
|
|
|
| 8 |
C |
0.224 |
|
|
|
| 9 |
N |
-0.683 |
|
|
|
| 10 |
H |
0.186 |
|
|
|
| 11 |
H |
0.197 |
|
|
|
| 12 |
H |
0.206 |
|
|
|
| 13 |
H |
0.372 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.221 |
2.992 |
0.000 |
3.726 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-49.576 |
6.190 |
0.000 |
| y |
6.190 |
-46.383 |
0.000 |
| z |
0.000 |
0.000 |
-51.247 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.761 |
6.190 |
0.000 |
| y |
6.190 |
4.029 |
0.000 |
| z |
0.000 |
0.000 |
-3.268 |
|
| Polar |
|---|
| 3z2-r2 | -6.536 |
|---|
| x2-y2 | -3.193 |
|---|
| xy | 6.190 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.554 |
1.333 |
0.000 |
| y |
1.333 |
11.287 |
0.000 |
| z |
0.000 |
0.000 |
4.213 |
<r2> (average value of r
2) Å
2
| <r2> |
252.536 |
| (<r2>)1/2 |
15.891 |