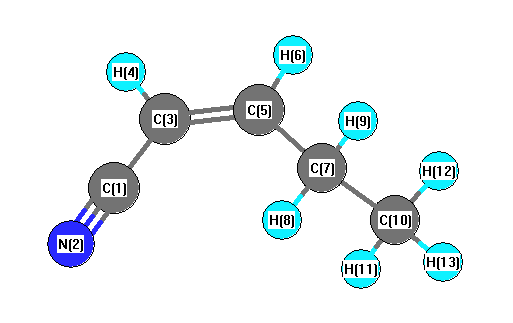
Vibrational Frequencies calculated at HSEh1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3228 |
3070 |
3.24 |
|
|
|
| 2 |
A |
3191 |
3034 |
10.47 |
|
|
|
| 3 |
A |
3165 |
3010 |
21.05 |
|
|
|
| 4 |
A |
3156 |
3001 |
23.46 |
|
|
|
| 5 |
A |
3124 |
2971 |
1.03 |
|
|
|
| 6 |
A |
3076 |
2925 |
27.17 |
|
|
|
| 7 |
A |
3057 |
2907 |
13.09 |
|
|
|
| 8 |
A |
2371 |
2254 |
19.59 |
|
|
|
| 9 |
A |
1723 |
1638 |
18.55 |
|
|
|
| 10 |
A |
1533 |
1458 |
7.38 |
|
|
|
| 11 |
A |
1526 |
1451 |
7.51 |
|
|
|
| 12 |
A |
1505 |
1432 |
5.70 |
|
|
|
| 13 |
A |
1442 |
1371 |
1.24 |
|
|
|
| 14 |
A |
1432 |
1362 |
0.83 |
|
|
|
| 15 |
A |
1354 |
1288 |
2.03 |
|
|
|
| 16 |
A |
1309 |
1244 |
0.01 |
|
|
|
| 17 |
A |
1267 |
1205 |
0.56 |
|
|
|
| 18 |
A |
1156 |
1099 |
0.83 |
|
|
|
| 19 |
A |
1104 |
1049 |
3.09 |
|
|
|
| 20 |
A |
1054 |
1002 |
4.47 |
|
|
|
| 21 |
A |
1004 |
955 |
1.03 |
|
|
|
| 22 |
A |
983 |
935 |
4.75 |
|
|
|
| 23 |
A |
884 |
840 |
4.41 |
|
|
|
| 24 |
A |
814 |
774 |
6.65 |
|
|
|
| 25 |
A |
772 |
734 |
37.80 |
|
|
|
| 26 |
A |
677 |
644 |
2.28 |
|
|
|
| 27 |
A |
580 |
552 |
0.74 |
|
|
|
| 28 |
A |
403 |
383 |
0.22 |
|
|
|
| 29 |
A |
370 |
352 |
0.04 |
|
|
|
| 30 |
A |
247 |
235 |
0.56 |
|
|
|
| 31 |
A |
215 |
205 |
3.14 |
|
|
|
| 32 |
A |
146 |
139 |
4.78 |
|
|
|
| 33 |
A |
59 |
57 |
1.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23962.5 cm
-1
Scaled (by 0.951) Zero Point Vibrational Energy (zpe) 22788.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.316 |
|
|
|
| 2 |
N |
-0.482 |
|
|
|
| 3 |
C |
-0.195 |
|
|
|
| 4 |
H |
0.215 |
|
|
|
| 5 |
C |
-0.078 |
|
|
|
| 6 |
H |
0.182 |
|
|
|
| 7 |
C |
-0.381 |
|
|
|
| 8 |
H |
0.204 |
|
|
|
| 9 |
H |
0.192 |
|
|
|
| 10 |
C |
-0.520 |
|
|
|
| 11 |
H |
0.189 |
|
|
|
| 12 |
H |
0.174 |
|
|
|
| 13 |
H |
0.185 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.371 |
2.511 |
0.051 |
4.204 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.999 |
-5.535 |
1.019 |
| y |
-5.535 |
-35.418 |
-0.708 |
| z |
1.019 |
-0.708 |
-36.521 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.030 |
-5.535 |
1.019 |
| y |
-5.535 |
3.842 |
-0.708 |
| z |
1.019 |
-0.708 |
2.188 |
|
| Polar |
|---|
| 3z2-r2 | 4.375 |
|---|
| x2-y2 | -6.581 |
|---|
| xy | -5.535 |
|---|
| xz | 1.019 |
|---|
| yz | -0.708 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.461 |
1.116 |
0.601 |
| y |
1.116 |
8.051 |
-0.312 |
| z |
0.601 |
-0.312 |
5.328 |
<r2> (average value of r
2) Å
2
| <r2> |
200.085 |
| (<r2>)1/2 |
14.145 |