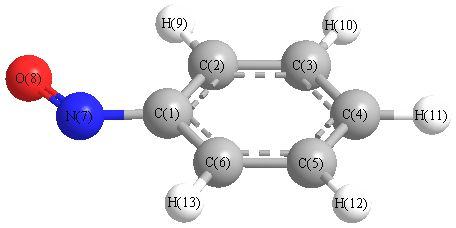
Vibrational Frequencies calculated at HSEh1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3250 |
3090 |
6.00 |
|
|
|
| 2 |
A' |
3244 |
3085 |
10.02 |
|
|
|
| 3 |
A' |
3235 |
3076 |
9.62 |
|
|
|
| 4 |
A' |
3228 |
3070 |
6.58 |
|
|
|
| 5 |
A' |
3216 |
3059 |
0.52 |
|
|
|
| 6 |
A' |
1702 |
1618 |
40.09 |
|
|
|
| 7 |
A' |
1678 |
1596 |
3.14 |
|
|
|
| 8 |
A' |
1639 |
1559 |
163.74 |
|
|
|
| 9 |
A' |
1524 |
1450 |
6.57 |
|
|
|
| 10 |
A' |
1510 |
1436 |
23.88 |
|
|
|
| 11 |
A' |
1409 |
1340 |
13.90 |
|
|
|
| 12 |
A' |
1343 |
1278 |
6.11 |
|
|
|
| 13 |
A' |
1215 |
1155 |
33.84 |
|
|
|
| 14 |
A' |
1195 |
1136 |
0.79 |
|
|
|
| 15 |
A' |
1155 |
1099 |
119.74 |
|
|
|
| 16 |
A' |
1109 |
1054 |
5.88 |
|
|
|
| 17 |
A' |
1052 |
1001 |
4.02 |
|
|
|
| 18 |
A' |
1023 |
973 |
0.51 |
|
|
|
| 19 |
A' |
848 |
807 |
28.86 |
|
|
|
| 20 |
A' |
687 |
654 |
9.45 |
|
|
|
| 21 |
A' |
623 |
593 |
0.06 |
|
|
|
| 22 |
A' |
452 |
430 |
0.58 |
|
|
|
| 23 |
A' |
259 |
246 |
2.49 |
|
|
|
| 24 |
A" |
1020 |
970 |
0.25 |
|
|
|
| 25 |
A" |
1000 |
951 |
0.05 |
|
|
|
| 26 |
A" |
966 |
919 |
4.16 |
|
|
|
| 27 |
A" |
874 |
831 |
0.00 |
|
|
|
| 28 |
A" |
782 |
744 |
55.46 |
|
|
|
| 29 |
A" |
698 |
664 |
20.60 |
|
|
|
| 30 |
A" |
475 |
451 |
1.33 |
|
|
|
| 31 |
A" |
421 |
400 |
0.00 |
|
|
|
| 32 |
A" |
251 |
239 |
0.25 |
|
|
|
| 33 |
A" |
120 |
114 |
0.16 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21600.5 cm
-1
Scaled (by 0.951) Zero Point Vibrational Energy (zpe) 20542.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.180 |
|
|
|
| 2 |
C |
-0.143 |
|
|
|
| 3 |
C |
-0.180 |
|
|
|
| 4 |
C |
-0.145 |
|
|
|
| 5 |
C |
-0.178 |
|
|
|
| 6 |
C |
-0.151 |
|
|
|
| 7 |
N |
-0.040 |
|
|
|
| 8 |
O |
-0.285 |
|
|
|
| 9 |
H |
0.201 |
|
|
|
| 10 |
H |
0.183 |
|
|
|
| 11 |
H |
0.182 |
|
|
|
| 12 |
H |
0.182 |
|
|
|
| 13 |
H |
0.194 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.166 |
-3.522 |
0.000 |
3.710 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.806 |
2.384 |
0.000 |
| y |
2.384 |
-47.420 |
0.000 |
| z |
0.000 |
0.000 |
-46.892 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.350 |
2.384 |
0.000 |
| y |
2.384 |
-3.570 |
0.000 |
| z |
0.000 |
0.000 |
-2.779 |
|
| Polar |
|---|
| 3z2-r2 | -5.558 |
|---|
| x2-y2 | 6.613 |
|---|
| xy | 2.384 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.029 |
-1.686 |
0.000 |
| y |
-1.686 |
14.555 |
0.000 |
| z |
0.000 |
0.000 |
3.775 |
<r2> (average value of r
2) Å
2
| <r2> |
247.772 |
| (<r2>)1/2 |
15.741 |