Vibrational Frequencies calculated at HSEh1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3302 |
3140 |
1.56 |
|
|
|
| 2 |
A |
3270 |
3110 |
0.95 |
|
|
|
| 3 |
A |
3173 |
3017 |
12.31 |
|
|
|
| 4 |
A |
3146 |
2992 |
11.09 |
|
|
|
| 5 |
A |
3077 |
2927 |
19.92 |
|
|
|
| 6 |
A |
1608 |
1529 |
35.01 |
|
|
|
| 7 |
A |
1524 |
1449 |
48.89 |
|
|
|
| 8 |
A |
1510 |
1436 |
2.94 |
|
|
|
| 9 |
A |
1503 |
1429 |
7.02 |
|
|
|
| 10 |
A |
1438 |
1368 |
4.20 |
|
|
|
| 11 |
A |
1385 |
1317 |
13.82 |
|
|
|
| 12 |
A |
1274 |
1212 |
1.03 |
|
|
|
| 13 |
A |
1181 |
1123 |
3.42 |
|
|
|
| 14 |
A |
1076 |
1024 |
5.45 |
|
|
|
| 15 |
A |
1028 |
977 |
9.50 |
|
|
|
| 16 |
A |
966 |
918 |
15.43 |
|
|
|
| 17 |
A |
906 |
862 |
29.30 |
|
|
|
| 18 |
A |
851 |
810 |
12.58 |
|
|
|
| 19 |
A |
825 |
785 |
41.49 |
|
|
|
| 20 |
A |
739 |
703 |
9.90 |
|
|
|
| 21 |
A |
686 |
652 |
1.45 |
|
|
|
| 22 |
A |
658 |
626 |
0.36 |
|
|
|
| 23 |
A |
565 |
538 |
1.82 |
|
|
|
| 24 |
A |
495 |
470 |
2.02 |
|
|
|
| 25 |
A |
334 |
318 |
2.49 |
|
|
|
| 26 |
A |
234 |
223 |
4.03 |
|
|
|
| 27 |
A |
128 |
121 |
0.49 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18440.8 cm
-1
Scaled (by 0.951) Zero Point Vibrational Energy (zpe) 17537.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
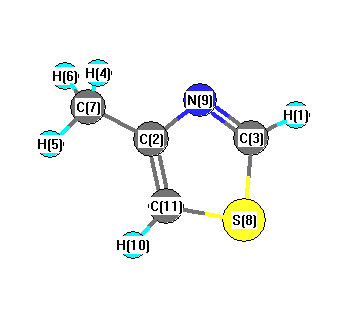
Charges from optimized geometry at HSEh1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.213 |
|
|
|
| 2 |
C |
0.292 |
|
|
|
| 3 |
C |
-0.172 |
|
|
|
| 4 |
H |
0.198 |
|
|
|
| 5 |
H |
0.178 |
|
|
|
| 6 |
H |
0.198 |
|
|
|
| 7 |
C |
-0.570 |
|
|
|
| 8 |
S |
0.276 |
|
|
|
| 9 |
N |
-0.384 |
|
|
|
| 10 |
H |
0.210 |
|
|
|
| 11 |
C |
-0.439 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.114 |
-1.167 |
0.000 |
1.173 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.707 |
-3.902 |
0.000 |
| y |
-3.902 |
-40.632 |
0.000 |
| z |
0.000 |
0.000 |
-43.924 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.571 |
-3.902 |
0.000 |
| y |
-3.902 |
0.684 |
0.000 |
| z |
0.000 |
0.000 |
-4.255 |
|
| Polar |
|---|
| 3z2-r2 | -8.510 |
|---|
| x2-y2 | 1.925 |
|---|
| xy | -3.902 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.611 |
0.253 |
0.000 |
| y |
0.253 |
9.368 |
0.000 |
| z |
0.000 |
0.000 |
4.904 |
<r2> (average value of r
2) Å
2
| <r2> |
178.583 |
| (<r2>)1/2 |
13.363 |