Jump to
S1C2
Energy calculated at HSEh1PBE/6-31G*
| | hartrees |
|---|
| Energy at 0K | -135.011928 |
| Energy at 298.15K | -135.020120 |
| Nuclear repulsion energy | 82.845341 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3498 |
3326 |
1.55 |
|
|
|
| 2 |
A' |
3137 |
2983 |
42.52 |
|
|
|
| 3 |
A' |
3070 |
2920 |
38.15 |
|
|
|
| 4 |
A' |
3055 |
2905 |
22.35 |
|
|
|
| 5 |
A' |
1709 |
1625 |
23.12 |
|
|
|
| 6 |
A' |
1532 |
1457 |
2.68 |
|
|
|
| 7 |
A' |
1513 |
1439 |
0.02 |
|
|
|
| 8 |
A' |
1435 |
1365 |
9.85 |
|
|
|
| 9 |
A' |
1399 |
1330 |
14.24 |
|
|
|
| 10 |
A' |
1181 |
1123 |
9.68 |
|
|
|
| 11 |
A' |
1096 |
1042 |
30.79 |
|
|
|
| 12 |
A' |
917 |
872 |
86.18 |
|
|
|
| 13 |
A' |
892 |
848 |
104.78 |
|
|
|
| 14 |
A' |
404 |
384 |
6.64 |
|
|
|
| 15 |
A" |
3587 |
3411 |
0.26 |
|
|
|
| 16 |
A" |
3138 |
2984 |
60.71 |
|
|
|
| 17 |
A" |
3106 |
2953 |
5.54 |
|
|
|
| 18 |
A" |
1523 |
1449 |
8.01 |
|
|
|
| 19 |
A" |
1414 |
1344 |
0.00 |
|
|
|
| 20 |
A" |
1289 |
1226 |
0.02 |
|
|
|
| 21 |
A" |
1020 |
970 |
1.55 |
|
|
|
| 22 |
A" |
784 |
746 |
1.55 |
|
|
|
| 23 |
A" |
310 |
295 |
46.99 |
|
|
|
| 24 |
A" |
261 |
248 |
11.47 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20633.5 cm
-1
Scaled (by 0.951) Zero Point Vibrational Energy (zpe) 19622.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.305 |
-0.077 |
0.000 |
| C2 |
0.000 |
0.572 |
0.000 |
| C3 |
1.208 |
-0.361 |
0.000 |
| H4 |
2.152 |
0.199 |
0.000 |
| H5 |
1.204 |
-1.008 |
0.886 |
| H6 |
1.204 |
-1.008 |
-0.886 |
| H7 |
0.042 |
1.232 |
-0.876 |
| H8 |
0.042 |
1.232 |
0.876 |
| H9 |
-1.380 |
-0.687 |
0.811 |
| H10 |
-1.380 |
-0.687 |
-0.811 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4574 | 2.5293 | 3.4676 | 2.8193 | 2.8193 | 2.0722 | 2.0722 | 1.0175 | 1.0175 |
C2 | 1.4574 | | 1.5263 | 2.1838 | 2.1753 | 2.1753 | 1.0972 | 1.0972 | 2.0360 | 2.0360 | C3 | 2.5293 | 1.5263 | | 1.0967 | 1.0971 | 1.0971 | 2.1591 | 2.1591 | 2.7317 | 2.7317 | H4 | 3.4676 | 2.1838 | 1.0967 | | 1.7715 | 1.7715 | 2.5067 | 2.5067 | 3.7299 | 3.7299 | H5 | 2.8193 | 2.1753 | 1.0971 | 1.7715 | | 1.7711 | 3.0773 | 2.5235 | 2.6051 | 3.1080 | H6 | 2.8193 | 2.1753 | 1.0971 | 1.7715 | 1.7711 | | 2.5235 | 3.0773 | 3.1080 | 2.6051 | H7 | 2.0722 | 1.0972 | 2.1591 | 2.5067 | 3.0773 | 2.5235 | | 1.7513 | 2.9235 | 2.3886 | H8 | 2.0722 | 1.0972 | 2.1591 | 2.5067 | 2.5235 | 3.0773 | 1.7513 | | 2.3886 | 2.9235 | H9 | 1.0175 | 2.0360 | 2.7317 | 3.7299 | 2.6051 | 3.1080 | 2.9235 | 2.3886 | | 1.6223 | H10 | 1.0175 | 2.0360 | 2.7317 | 3.7299 | 3.1080 | 2.6051 | 2.3886 | 2.9235 | 1.6223 | |
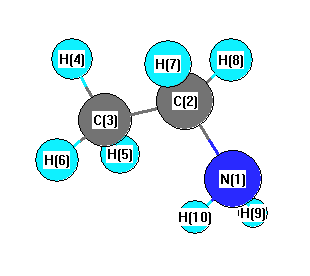 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
115.907 |
|
N1 |
C2 |
H7 |
107.587 |
| N1 |
C2 |
H8 |
107.587 |
|
C2 |
N1 |
H9 |
109.420 |
| C2 |
N1 |
H10 |
109.420 |
|
C2 |
C3 |
H4 |
111.678 |
| C2 |
C3 |
H5 |
110.965 |
|
C2 |
C3 |
H6 |
110.965 |
| C3 |
C2 |
H7 |
109.688 |
|
C3 |
C2 |
H8 |
109.688 |
| H4 |
C3 |
H5 |
107.708 |
|
H4 |
C3 |
H6 |
107.708 |
| H5 |
C3 |
H6 |
107.638 |
|
H7 |
C2 |
H8 |
105.896 |
| H9 |
N1 |
H10 |
105.729 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.734 |
|
|
|
| 2 |
C |
-0.198 |
|
|
|
| 3 |
C |
-0.520 |
|
|
|
| 4 |
H |
0.167 |
|
|
|
| 5 |
H |
0.162 |
|
|
|
| 6 |
H |
0.162 |
|
|
|
| 7 |
H |
0.171 |
|
|
|
| 8 |
H |
0.171 |
|
|
|
| 9 |
H |
0.309 |
|
|
|
| 10 |
H |
0.309 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.989 |
-1.084 |
0.000 |
1.467 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.830 |
2.695 |
0.000 |
| y |
2.695 |
-19.964 |
0.000 |
| z |
0.000 |
0.000 |
-18.894 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.401 |
2.695 |
0.000 |
| y |
2.695 |
1.398 |
0.000 |
| z |
0.000 |
0.000 |
3.003 |
|
| Polar |
|---|
| 3z2-r2 | 6.006 |
|---|
| x2-y2 | -3.867 |
|---|
| xy | 2.695 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.664 |
0.255 |
0.000 |
| y |
0.255 |
4.237 |
0.000 |
| z |
0.000 |
0.000 |
4.302 |
<r2> (average value of r
2) Å
2
| <r2> |
58.326 |
| (<r2>)1/2 |
7.637 |
Jump to
S1C1
Energy calculated at HSEh1PBE/6-31G*
| | hartrees |
|---|
| Energy at 0K | -135.011286 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-31G*
Geometric Data calculated at HSEh1PBE/6-31G*
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability