Jump to
S1C2
S1C3
Energy calculated at HSEh1PBE/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.262640 |
| Energy at 298.15K | -100.261705 |
| HF Energy | -100.262640 |
| Nuclear repulsion energy | 27.522784 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
2243 |
2158 |
7.29 |
|
|
|
| 2 |
Σ |
622 |
598 |
127.34 |
|
|
|
| 3 |
Π |
171 |
165 |
34.11 |
|
|
|
| 3 |
Π |
171 |
165 |
34.12 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1603.4 cm
-1
Scaled (by 0.9622) Zero Point Vibrational Energy (zpe) 1542.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/aug-cc-pVDZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.078 |
| C2 |
0.000 |
0.000 |
-0.150 |
| N3 |
0.000 |
0.000 |
1.020 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9278 | 3.0979 |
C2 | 1.9278 | | 1.1701 | N3 | 3.0979 | 1.1701 | |
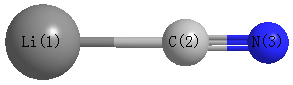 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Li |
0.146 |
|
|
|
| 2 |
C |
0.301 |
|
|
|
| 3 |
N |
-0.447 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-9.279 |
9.279 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-14.452 |
0.000 |
0.000 |
| y |
0.000 |
-14.452 |
0.000 |
| z |
0.000 |
0.000 |
1.210 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.831 |
0.000 |
0.000 |
| y |
0.000 |
-7.831 |
0.000 |
| z |
0.000 |
0.000 |
15.663 |
|
| Polar |
|---|
| 3z2-r2 | 31.325 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.116 |
0.000 |
0.000 |
| y |
0.000 |
3.116 |
0.000 |
| z |
0.000 |
0.000 |
4.761 |
<r2> (average value of r
2) Å
2
| <r2> |
26.137 |
| (<r2>)1/2 |
5.112 |
Jump to
S1C1
S1C3
Energy calculated at HSEh1PBE/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.265670 |
| Energy at 298.15K | -100.264873 |
| HF Energy | -100.265670 |
| Nuclear repulsion energy | 29.124934 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at HSEh1PBE/aug-cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.442 |
-0.585 |
0.000 |
| C2 |
-0.721 |
-0.370 |
0.000 |
| N3 |
0.000 |
0.568 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.1742 | 1.8461 |
C2 | 2.1742 | | 1.1827 | N3 | 1.8461 | 1.1827 | |
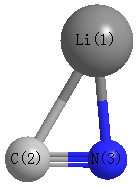 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
58.100 |
|
Li1 |
N3 |
C2 |
88.952 |
| C2 |
Li1 |
N3 |
32.948 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Li |
0.459 |
|
|
|
| 2 |
C |
-0.172 |
|
|
|
| 3 |
N |
-0.288 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
6.716 |
-2.389 |
0.000 |
7.128 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-5.810 |
-5.940 |
0.000 |
| y |
-5.940 |
-15.205 |
0.000 |
| z |
0.000 |
0.000 |
-14.583 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
9.084 |
-5.940 |
0.000 |
| y |
-5.940 |
-5.009 |
0.000 |
| z |
0.000 |
0.000 |
-4.075 |
|
| Polar |
|---|
| 3z2-r2 | -8.150 |
|---|
| x2-y2 | 9.396 |
|---|
| xy | -5.940 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.898 |
0.373 |
0.000 |
| y |
0.373 |
3.802 |
0.000 |
| z |
0.000 |
0.000 |
3.198 |
<r2> (average value of r
2) Å
2
| <r2> |
21.003 |
| (<r2>)1/2 |
4.583 |
Jump to
S1C1
S1C2
Energy calculated at HSEh1PBE/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.267022 |
| Energy at 298.15K | -100.265929 |
| HF Energy | -100.267022 |
| Nuclear repulsion energy | 28.268761 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
2160 |
2079 |
173.84 |
|
|
|
| 2 |
Σ |
711 |
684 |
164.12 |
|
|
|
| 3 |
Π |
110 |
106 |
26.15 |
|
|
|
| 3 |
Π |
110 |
106 |
26.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1545.8 cm
-1
Scaled (by 0.9622) Zero Point Vibrational Energy (zpe) 1487.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/aug-cc-pVDZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.891 |
| C2 |
0.000 |
0.000 |
-1.072 |
| N3 |
0.000 |
0.000 |
0.109 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9628 | 1.7817 |
C2 | 2.9628 | | 1.1812 | N3 | 1.7817 | 1.1812 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Li |
0.380 |
|
|
|
| 2 |
C |
-0.488 |
|
|
|
| 3 |
N |
0.108 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
9.013 |
9.013 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-14.457 |
0.000 |
0.000 |
| y |
0.000 |
-14.457 |
0.000 |
| z |
0.000 |
0.000 |
-2.305 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.076 |
0.000 |
0.000 |
| y |
0.000 |
-6.076 |
0.000 |
| z |
0.000 |
0.000 |
12.153 |
|
| Polar |
|---|
| 3z2-r2 | 24.305 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.163 |
0.000 |
-0.000 |
| y |
0.000 |
3.163 |
0.000 |
| z |
-0.000 |
0.000 |
4.827 |
<r2> (average value of r
2) Å
2
| <r2> |
24.204 |
| (<r2>)1/2 |
4.920 |