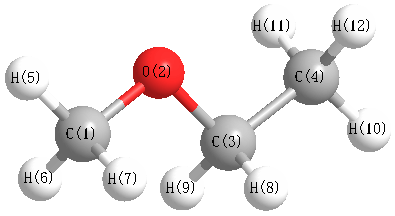
Vibrational Frequencies calculated at HSEh1PBE/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3132 |
3012 |
21.92 |
|
|
|
| 2 |
A' |
3131 |
3011 |
24.65 |
|
|
|
| 3 |
A' |
3055 |
2938 |
16.31 |
|
|
|
| 4 |
A' |
2983 |
2869 |
85.33 |
|
|
|
| 5 |
A' |
2966 |
2853 |
40.53 |
|
|
|
| 6 |
A' |
1521 |
1463 |
3.60 |
|
|
|
| 7 |
A' |
1499 |
1441 |
3.69 |
|
|
|
| 8 |
A' |
1492 |
1435 |
6.16 |
|
|
|
| 9 |
A' |
1470 |
1414 |
0.86 |
|
|
|
| 10 |
A' |
1422 |
1368 |
29.34 |
|
|
|
| 11 |
A' |
1389 |
1336 |
0.49 |
|
|
|
| 12 |
A' |
1240 |
1193 |
54.72 |
|
|
|
| 13 |
A' |
1185 |
1140 |
140.25 |
|
|
|
| 14 |
A' |
1118 |
1075 |
4.23 |
|
|
|
| 15 |
A' |
1053 |
1013 |
19.30 |
|
|
|
| 16 |
A' |
877 |
843 |
9.57 |
|
|
|
| 17 |
A' |
468 |
450 |
0.58 |
|
|
|
| 18 |
A' |
288 |
277 |
2.69 |
|
|
|
| 19 |
A" |
3134 |
3014 |
24.12 |
|
|
|
| 20 |
A" |
3028 |
2913 |
51.79 |
|
|
|
| 21 |
A" |
2992 |
2878 |
63.86 |
|
|
|
| 22 |
A" |
1481 |
1425 |
9.75 |
|
|
|
| 23 |
A" |
1475 |
1419 |
4.93 |
|
|
|
| 24 |
A" |
1299 |
1250 |
1.89 |
|
|
|
| 25 |
A" |
1195 |
1149 |
8.96 |
|
|
|
| 26 |
A" |
1165 |
1121 |
0.10 |
|
|
|
| 27 |
A" |
820 |
789 |
0.56 |
|
|
|
| 28 |
A" |
257 |
247 |
1.71 |
|
|
|
| 29 |
A" |
207 |
199 |
1.05 |
|
|
|
| 30 |
A" |
117 |
113 |
2.59 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23729.3 cm
-1
Scaled (by 0.9618) Zero Point Vibrational Energy (zpe) 22822.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.648 |
|
|
|
| 2 |
O |
-0.453 |
|
|
|
| 3 |
C |
-0.026 |
|
|
|
| 4 |
C |
-0.707 |
|
|
|
| 5 |
H |
0.259 |
|
|
|
| 6 |
H |
0.256 |
|
|
|
| 7 |
H |
0.256 |
|
|
|
| 8 |
H |
0.209 |
|
|
|
| 9 |
H |
0.209 |
|
|
|
| 10 |
H |
0.228 |
|
|
|
| 11 |
H |
0.208 |
|
|
|
| 12 |
H |
0.208 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.887 |
-0.724 |
0.000 |
1.145 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.954 |
1.735 |
0.000 |
| y |
1.735 |
-25.851 |
0.000 |
| z |
0.000 |
0.000 |
-26.620 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.282 |
1.735 |
0.000 |
| y |
1.735 |
0.436 |
0.000 |
| z |
0.000 |
0.000 |
-0.718 |
|
| Polar |
|---|
| 3z2-r2 | -1.436 |
|---|
| x2-y2 | -0.102 |
|---|
| xy | 1.735 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.330 |
0.827 |
0.000 |
| y |
0.827 |
7.181 |
0.000 |
| z |
0.000 |
0.000 |
6.171 |
<r2> (average value of r
2) Å
2
| <r2> |
102.727 |
| (<r2>)1/2 |
10.135 |