Vibrational Frequencies calculated at HSEh1PBE/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3250 |
3126 |
4.53 |
|
|
|
| 2 |
A' |
3186 |
3064 |
3.67 |
|
|
|
| 3 |
A' |
3170 |
3049 |
4.55 |
|
|
|
| 4 |
A' |
3156 |
3036 |
4.71 |
|
|
|
| 5 |
A' |
3138 |
3019 |
4.35 |
|
|
|
| 6 |
A' |
2889 |
2778 |
88.51 |
|
|
|
| 7 |
A' |
1787 |
1719 |
412.42 |
|
|
|
| 8 |
A' |
1709 |
1644 |
74.58 |
|
|
|
| 9 |
A' |
1662 |
1599 |
31.61 |
|
|
|
| 10 |
A' |
1453 |
1398 |
5.14 |
|
|
|
| 11 |
A' |
1407 |
1353 |
0.02 |
|
|
|
| 12 |
A' |
1322 |
1271 |
2.52 |
|
|
|
| 13 |
A' |
1317 |
1267 |
2.45 |
|
|
|
| 14 |
A' |
1260 |
1212 |
4.46 |
|
|
|
| 15 |
A' |
1198 |
1152 |
30.49 |
|
|
|
| 16 |
A' |
1137 |
1094 |
111.85 |
|
|
|
| 17 |
A' |
966 |
929 |
5.34 |
|
|
|
| 18 |
A' |
603 |
580 |
16.15 |
|
|
|
| 19 |
A' |
434 |
418 |
0.53 |
|
|
|
| 20 |
A' |
383 |
368 |
4.06 |
|
|
|
| 21 |
A' |
148 |
142 |
6.41 |
|
|
|
| 22 |
A" |
1054 |
1014 |
44.73 |
|
|
|
| 23 |
A" |
1030 |
991 |
4.38 |
|
|
|
| 24 |
A" |
996 |
958 |
22.70 |
|
|
|
| 25 |
A" |
968 |
931 |
21.24 |
|
|
|
| 26 |
A" |
890 |
856 |
8.79 |
|
|
|
| 27 |
A" |
659 |
634 |
2.76 |
|
|
|
| 28 |
A" |
287 |
276 |
7.30 |
|
|
|
| 29 |
A" |
214 |
205 |
1.73 |
|
|
|
| 30 |
A" |
96 |
93 |
2.99 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20884.4 cm
-1
Scaled (by 0.9618) Zero Point Vibrational Energy (zpe) 20086.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
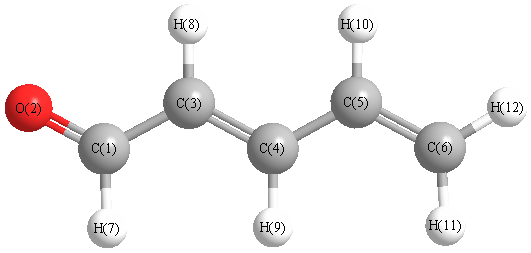
Charges from optimized geometry at HSEh1PBE/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.026 |
|
|
|
| 2 |
O |
-0.648 |
|
|
|
| 3 |
C |
-0.281 |
|
|
|
| 4 |
C |
0.091 |
|
|
|
| 5 |
C |
-0.041 |
|
|
|
| 6 |
C |
-0.945 |
|
|
|
| 7 |
H |
0.382 |
|
|
|
| 8 |
H |
0.353 |
|
|
|
| 9 |
H |
0.231 |
|
|
|
| 10 |
H |
0.293 |
|
|
|
| 11 |
H |
0.194 |
|
|
|
| 12 |
H |
0.397 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.032 |
4.128 |
0.000 |
4.255 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.918 |
-3.193 |
0.000 |
| y |
-3.193 |
-43.425 |
0.000 |
| z |
0.000 |
0.000 |
-38.234 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.912 |
-3.193 |
0.000 |
| y |
-3.193 |
-7.849 |
0.000 |
| z |
0.000 |
0.000 |
-0.062 |
|
| Polar |
|---|
| 3z2-r2 | -0.124 |
|---|
| x2-y2 | 10.507 |
|---|
| xy | -3.193 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.618 |
3.065 |
0.002 |
| y |
3.065 |
18.703 |
0.003 |
| z |
0.002 |
0.003 |
6.243 |
<r2> (average value of r
2) Å
2
| <r2> |
240.090 |
| (<r2>)1/2 |
15.495 |