Vibrational Frequencies calculated at HSEh1PBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3248 |
3129 |
9.43 |
|
|
|
| 2 |
A' |
3241 |
3122 |
5.70 |
|
|
|
| 3 |
A' |
3232 |
3114 |
9.57 |
|
|
|
| 4 |
A' |
3223 |
3105 |
4.13 |
|
|
|
| 5 |
A' |
3214 |
3096 |
0.03 |
|
|
|
| 6 |
A' |
3192 |
3075 |
7.71 |
|
|
|
| 7 |
A' |
3074 |
2961 |
1.22 |
|
|
|
| 8 |
A' |
1749 |
1685 |
92.24 |
|
|
|
| 9 |
A' |
1655 |
1595 |
16.10 |
|
|
|
| 10 |
A' |
1635 |
1575 |
9.06 |
|
|
|
| 11 |
A' |
1549 |
1493 |
0.33 |
|
|
|
| 12 |
A' |
1520 |
1464 |
14.31 |
|
|
|
| 13 |
A' |
1511 |
1455 |
19.95 |
|
|
|
| 14 |
A' |
1428 |
1376 |
54.02 |
|
|
|
| 15 |
A' |
1385 |
1334 |
4.77 |
|
|
|
| 16 |
A' |
1362 |
1312 |
1.87 |
|
|
|
| 17 |
A' |
1296 |
1249 |
189.47 |
|
|
|
| 18 |
A' |
1241 |
1196 |
21.36 |
|
|
|
| 19 |
A' |
1231 |
1185 |
9.32 |
|
|
|
| 20 |
A' |
1130 |
1089 |
9.66 |
|
|
|
| 21 |
A' |
1122 |
1081 |
1.68 |
|
|
|
| 22 |
A' |
1068 |
1029 |
8.06 |
|
|
|
| 23 |
A' |
1047 |
1008 |
0.00 |
|
|
|
| 24 |
A' |
977 |
941 |
27.87 |
|
|
|
| 25 |
A' |
759 |
731 |
0.08 |
|
|
|
| 26 |
A' |
659 |
635 |
0.68 |
|
|
|
| 27 |
A' |
596 |
574 |
21.72 |
|
|
|
| 28 |
A' |
480 |
463 |
0.69 |
|
|
|
| 29 |
A' |
376 |
363 |
1.48 |
|
|
|
| 30 |
A' |
226 |
218 |
5.43 |
|
|
|
| 31 |
A" |
3142 |
3026 |
3.58 |
|
|
|
| 32 |
A" |
1544 |
1487 |
16.44 |
|
|
|
| 33 |
A" |
1098 |
1058 |
3.11 |
|
|
|
| 34 |
A" |
1058 |
1019 |
0.95 |
|
|
|
| 35 |
A" |
1034 |
996 |
0.74 |
|
|
|
| 36 |
A" |
978 |
942 |
3.96 |
|
|
|
| 37 |
A" |
888 |
856 |
0.33 |
|
|
|
| 38 |
A" |
815 |
785 |
25.40 |
|
|
|
| 39 |
A" |
738 |
711 |
69.82 |
|
|
|
| 40 |
A" |
639 |
615 |
12.68 |
|
|
|
| 41 |
A" |
442 |
426 |
1.33 |
|
|
|
| 42 |
A" |
430 |
414 |
0.02 |
|
|
|
| 43 |
A" |
203 |
196 |
1.05 |
|
|
|
| 44 |
A" |
160 |
154 |
0.03 |
|
|
|
| 45 |
A" |
85 |
81 |
3.08 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30839.9 cm
-1
Scaled (by 0.9633) Zero Point Vibrational Energy (zpe) 29708.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
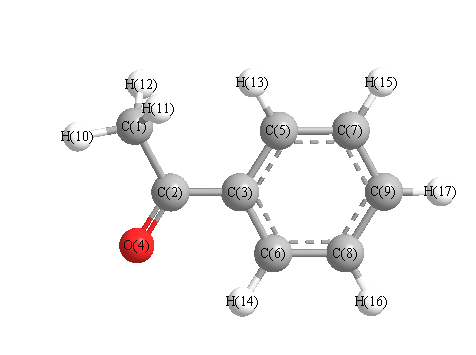
Charges from optimized geometry at HSEh1PBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.708 |
|
|
|
| 2 |
C |
0.437 |
|
|
|
| 3 |
C |
-0.113 |
|
|
|
| 4 |
O |
-0.463 |
|
|
|
| 5 |
C |
-0.223 |
|
|
|
| 6 |
C |
-0.178 |
|
|
|
| 7 |
C |
-0.214 |
|
|
|
| 8 |
C |
-0.220 |
|
|
|
| 9 |
C |
-0.209 |
|
|
|
| 10 |
H |
0.254 |
|
|
|
| 11 |
H |
0.243 |
|
|
|
| 12 |
H |
0.243 |
|
|
|
| 13 |
H |
0.221 |
|
|
|
| 14 |
H |
0.250 |
|
|
|
| 15 |
H |
0.225 |
|
|
|
| 16 |
H |
0.227 |
|
|
|
| 17 |
H |
0.227 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.005 |
-2.127 |
0.000 |
2.923 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.166 |
5.670 |
0.000 |
| y |
5.670 |
-51.714 |
0.000 |
| z |
0.000 |
0.000 |
-55.975 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.679 |
5.670 |
0.000 |
| y |
5.670 |
-0.143 |
0.000 |
| z |
0.000 |
0.000 |
-6.535 |
|
| Polar |
|---|
| 3z2-r2 | -13.071 |
|---|
| x2-y2 | 4.548 |
|---|
| xy | 5.670 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.759 |
0.376 |
0.000 |
| y |
0.376 |
15.702 |
0.000 |
| z |
0.000 |
0.000 |
4.242 |
<r2> (average value of r
2) Å
2
| <r2> |
342.924 |
| (<r2>)1/2 |
18.518 |