Jump to
S1C2
Energy calculated at HSEh1PBE/6-311G*
| | hartrees |
|---|
| Energy at 0K | -311.998258 |
| Energy at 298.15K | -312.013821 |
| HF Energy | -311.998258 |
| Nuclear repulsion energy | 315.698394 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3128 |
3002 |
53.11 |
|
|
|
| 2 |
A1 |
3061 |
2937 |
59.24 |
|
|
|
| 3 |
A1 |
3047 |
2924 |
20.17 |
|
|
|
| 4 |
A1 |
2975 |
2855 |
102.37 |
|
|
|
| 5 |
A1 |
1548 |
1486 |
3.85 |
|
|
|
| 6 |
A1 |
1522 |
1461 |
9.66 |
|
|
|
| 7 |
A1 |
1506 |
1445 |
0.34 |
|
|
|
| 8 |
A1 |
1472 |
1412 |
0.12 |
|
|
|
| 9 |
A1 |
1426 |
1368 |
3.01 |
|
|
|
| 10 |
A1 |
1348 |
1294 |
2.42 |
|
|
|
| 11 |
A1 |
1190 |
1142 |
2.75 |
|
|
|
| 12 |
A1 |
1086 |
1042 |
8.96 |
|
|
|
| 13 |
A1 |
1031 |
989 |
18.76 |
|
|
|
| 14 |
A1 |
934 |
896 |
8.59 |
|
|
|
| 15 |
A1 |
439 |
421 |
0.51 |
|
|
|
| 16 |
A1 |
323 |
310 |
0.27 |
|
|
|
| 17 |
A1 |
105 |
101 |
0.12 |
|
|
|
| 18 |
A2 |
3120 |
2994 |
0.00 |
|
|
|
| 19 |
A2 |
3093 |
2968 |
0.00 |
|
|
|
| 20 |
A2 |
2997 |
2875 |
0.00 |
|
|
|
| 21 |
A2 |
1516 |
1455 |
0.00 |
|
|
|
| 22 |
A2 |
1331 |
1277 |
0.00 |
|
|
|
| 23 |
A2 |
1281 |
1229 |
0.00 |
|
|
|
| 24 |
A2 |
1187 |
1139 |
0.00 |
|
|
|
| 25 |
A2 |
902 |
866 |
0.00 |
|
|
|
| 26 |
A2 |
768 |
737 |
0.00 |
|
|
|
| 27 |
A2 |
234 |
225 |
0.00 |
|
|
|
| 28 |
A2 |
140 |
135 |
0.00 |
|
|
|
| 29 |
A2 |
75 |
72 |
0.00 |
|
|
|
| 30 |
B1 |
3120 |
2994 |
148.57 |
|
|
|
| 31 |
B1 |
3093 |
2968 |
4.58 |
|
|
|
| 32 |
B1 |
2994 |
2873 |
124.38 |
|
|
|
| 33 |
B1 |
1516 |
1455 |
20.05 |
|
|
|
| 34 |
B1 |
1335 |
1281 |
1.36 |
|
|
|
| 35 |
B1 |
1290 |
1238 |
3.97 |
|
|
|
| 36 |
B1 |
1209 |
1160 |
4.49 |
|
|
|
| 37 |
B1 |
917 |
879 |
4.03 |
|
|
|
| 38 |
B1 |
771 |
740 |
4.10 |
|
|
|
| 39 |
B1 |
238 |
228 |
0.31 |
|
|
|
| 40 |
B1 |
161 |
155 |
5.27 |
|
|
|
| 41 |
B1 |
58 |
56 |
0.22 |
|
|
|
| 42 |
B2 |
3128 |
3001 |
25.05 |
|
|
|
| 43 |
B2 |
3060 |
2936 |
4.18 |
|
|
|
| 44 |
B2 |
3047 |
2924 |
44.10 |
|
|
|
| 45 |
B2 |
2963 |
2843 |
13.69 |
|
|
|
| 46 |
B2 |
1530 |
1469 |
12.58 |
|
|
|
| 47 |
B2 |
1519 |
1458 |
0.80 |
|
|
|
| 48 |
B2 |
1504 |
1443 |
1.56 |
|
|
|
| 49 |
B2 |
1426 |
1368 |
0.67 |
|
|
|
| 50 |
B2 |
1423 |
1365 |
48.44 |
|
|
|
| 51 |
B2 |
1335 |
1281 |
6.57 |
|
|
|
| 52 |
B2 |
1189 |
1141 |
251.91 |
|
|
|
| 53 |
B2 |
1139 |
1093 |
3.75 |
|
|
|
| 54 |
B2 |
1073 |
1029 |
0.17 |
|
|
|
| 55 |
B2 |
910 |
874 |
2.22 |
|
|
|
| 56 |
B2 |
520 |
499 |
5.77 |
|
|
|
| 57 |
B2 |
252 |
242 |
0.75 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42752.4 cm
-1
Scaled (by 0.9596) Zero Point Vibrational Energy (zpe) 41025.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-311G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.386 |
| C2 |
0.000 |
1.172 |
-0.390 |
| C3 |
0.000 |
-1.172 |
-0.390 |
| C4 |
0.000 |
2.372 |
0.531 |
| C5 |
0.000 |
-2.372 |
0.531 |
| C6 |
0.000 |
3.689 |
-0.230 |
| C7 |
0.000 |
-3.689 |
-0.230 |
| H8 |
0.886 |
1.193 |
-1.047 |
| H9 |
-0.886 |
1.193 |
-1.047 |
| H10 |
-0.886 |
-1.193 |
-1.047 |
| H11 |
0.886 |
-1.193 |
-1.047 |
| H12 |
0.877 |
2.306 |
1.184 |
| H13 |
-0.877 |
2.306 |
1.184 |
| H14 |
-0.877 |
-2.306 |
1.184 |
| H15 |
0.877 |
-2.306 |
1.184 |
| H16 |
0.000 |
4.542 |
0.453 |
| H17 |
-0.882 |
3.784 |
-0.871 |
| H18 |
0.882 |
3.784 |
-0.871 |
| H19 |
0.000 |
-4.542 |
0.453 |
| H20 |
0.882 |
-3.784 |
-0.871 |
| H21 |
-0.882 |
-3.784 |
-0.871 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4056 | 1.4056 | 2.3767 | 2.3767 | 3.7402 | 3.7402 | 2.0644 | 2.0644 | 2.0644 | 2.0644 | 2.5932 | 2.5932 | 2.5932 | 2.5932 | 4.5425 | 4.0834 | 4.0834 | 4.5425 | 4.0834 | 4.0834 |
C2 | 1.4056 | | 2.3440 | 1.5128 | 3.6620 | 2.5221 | 4.8637 | 1.1033 | 1.1033 | 2.6098 | 2.6098 | 2.1289 | 2.1289 | 3.9172 | 3.9172 | 3.4738 | 2.7983 | 2.7983 | 5.7759 | 5.0565 | 5.0565 | C3 | 1.4056 | 2.3440 | | 3.6620 | 1.5128 | 4.8637 | 2.5221 | 2.6098 | 2.6098 | 1.1033 | 1.1033 | 3.9172 | 3.9172 | 2.1289 | 2.1289 | 5.7759 | 5.0565 | 5.0565 | 3.4738 | 2.7983 | 2.7983 | C4 | 2.3767 | 1.5128 | 3.6620 | | 4.7445 | 1.5209 | 6.1089 | 2.1598 | 2.1598 | 3.9984 | 3.9984 | 1.0952 | 1.0952 | 4.8047 | 4.8047 | 2.1712 | 2.1762 | 2.1762 | 6.9147 | 6.3749 | 6.3749 | C5 | 2.3767 | 3.6620 | 1.5128 | 4.7445 | | 6.1089 | 1.5209 | 3.9984 | 3.9984 | 2.1598 | 2.1598 | 4.8047 | 4.8047 | 1.0952 | 1.0952 | 6.9147 | 6.3749 | 6.3749 | 2.1712 | 2.1762 | 2.1762 | C6 | 3.7402 | 2.5221 | 4.8637 | 1.5209 | 6.1089 | | 7.3781 | 2.7716 | 2.7716 | 5.0288 | 5.0288 | 2.1632 | 2.1632 | 6.2220 | 6.2220 | 1.0927 | 1.0946 | 1.0946 | 8.2594 | 7.5519 | 7.5519 | C7 | 3.7402 | 4.8637 | 2.5221 | 6.1089 | 1.5209 | 7.3781 | | 5.0288 | 5.0288 | 2.7716 | 2.7716 | 6.2220 | 6.2220 | 2.1632 | 2.1632 | 8.2594 | 7.5519 | 7.5519 | 1.0927 | 1.0946 | 1.0946 | H8 | 2.0644 | 1.1033 | 2.6098 | 2.1598 | 3.9984 | 2.7716 | 5.0288 | | 1.7724 | 2.9726 | 2.3864 | 2.4929 | 3.0533 | 4.5090 | 4.1500 | 3.7748 | 3.1415 | 2.5964 | 5.9939 | 4.9800 | 5.2847 | H9 | 2.0644 | 1.1033 | 2.6098 | 2.1598 | 3.9984 | 2.7716 | 5.0288 | 1.7724 | | 2.3864 | 2.9726 | 3.0533 | 2.4929 | 4.1500 | 4.5090 | 3.7748 | 2.5964 | 3.1415 | 5.9939 | 5.2847 | 4.9800 | H10 | 2.0644 | 2.6098 | 1.1033 | 3.9984 | 2.1598 | 5.0288 | 2.7716 | 2.9726 | 2.3864 | | 1.7724 | 4.5090 | 4.1500 | 2.4929 | 3.0533 | 5.9939 | 4.9800 | 5.2847 | 3.7748 | 3.1415 | 2.5964 | H11 | 2.0644 | 2.6098 | 1.1033 | 3.9984 | 2.1598 | 5.0288 | 2.7716 | 2.3864 | 2.9726 | 1.7724 | | 4.1500 | 4.5090 | 3.0533 | 2.4929 | 5.9939 | 5.2847 | 4.9800 | 3.7748 | 2.5964 | 3.1415 | H12 | 2.5932 | 2.1289 | 3.9172 | 1.0952 | 4.8047 | 2.1632 | 6.2220 | 2.4929 | 3.0533 | 4.5090 | 4.1500 | | 1.7539 | 4.9350 | 4.6128 | 2.5102 | 3.0820 | 2.5305 | 6.9429 | 6.4273 | 6.6637 | H13 | 2.5932 | 2.1289 | 3.9172 | 1.0952 | 4.8047 | 2.1632 | 6.2220 | 3.0533 | 2.4929 | 4.1500 | 4.5090 | 1.7539 | | 4.6128 | 4.9350 | 2.5102 | 2.5305 | 3.0820 | 6.9429 | 6.6637 | 6.4273 | H14 | 2.5932 | 3.9172 | 2.1289 | 4.8047 | 1.0952 | 6.2220 | 2.1632 | 4.5090 | 4.1500 | 2.4929 | 3.0533 | 4.9350 | 4.6128 | | 1.7539 | 6.9429 | 6.4273 | 6.6637 | 2.5102 | 3.0820 | 2.5305 | H15 | 2.5932 | 3.9172 | 2.1289 | 4.8047 | 1.0952 | 6.2220 | 2.1632 | 4.1500 | 4.5090 | 3.0533 | 2.4929 | 4.6128 | 4.9350 | 1.7539 | | 6.9429 | 6.6637 | 6.4273 | 2.5102 | 2.5305 | 3.0820 | H16 | 4.5425 | 3.4738 | 5.7759 | 2.1712 | 6.9147 | 1.0927 | 8.2594 | 3.7748 | 3.7748 | 5.9939 | 5.9939 | 2.5102 | 2.5102 | 6.9429 | 6.9429 | | 1.7624 | 1.7624 | 9.0840 | 8.4763 | 8.4763 | H17 | 4.0834 | 2.7983 | 5.0565 | 2.1762 | 6.3749 | 1.0946 | 7.5519 | 3.1415 | 2.5964 | 4.9800 | 5.2847 | 3.0820 | 2.5305 | 6.4273 | 6.6637 | 1.7624 | | 1.7648 | 8.4763 | 7.7703 | 7.5673 | H18 | 4.0834 | 2.7983 | 5.0565 | 2.1762 | 6.3749 | 1.0946 | 7.5519 | 2.5964 | 3.1415 | 5.2847 | 4.9800 | 2.5305 | 3.0820 | 6.6637 | 6.4273 | 1.7624 | 1.7648 | | 8.4763 | 7.5673 | 7.7703 | H19 | 4.5425 | 5.7759 | 3.4738 | 6.9147 | 2.1712 | 8.2594 | 1.0927 | 5.9939 | 5.9939 | 3.7748 | 3.7748 | 6.9429 | 6.9429 | 2.5102 | 2.5102 | 9.0840 | 8.4763 | 8.4763 | | 1.7624 | 1.7624 | H20 | 4.0834 | 5.0565 | 2.7983 | 6.3749 | 2.1762 | 7.5519 | 1.0946 | 4.9800 | 5.2847 | 3.1415 | 2.5964 | 6.4273 | 6.6637 | 3.0820 | 2.5305 | 8.4763 | 7.7703 | 7.5673 | 1.7624 | | 1.7648 | H21 | 4.0834 | 5.0565 | 2.7983 | 6.3749 | 2.1762 | 7.5519 | 1.0946 | 5.2847 | 4.9800 | 2.5964 | 3.1415 | 6.6637 | 6.4273 | 2.5305 | 3.0820 | 8.4763 | 7.5673 | 7.7703 | 1.7624 | 1.7648 | |
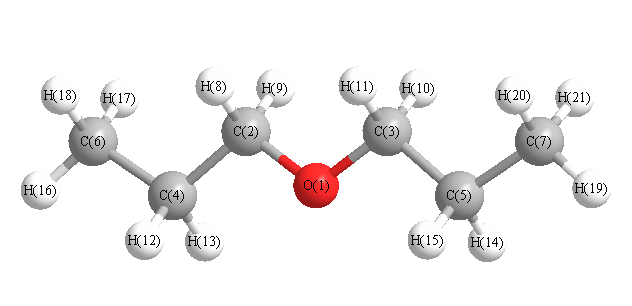 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.997 |
|
O1 |
C2 |
H8 |
110.162 |
| O1 |
C2 |
H9 |
110.162 |
|
O1 |
C3 |
C5 |
108.997 |
| O1 |
C3 |
H10 |
110.162 |
|
O1 |
C3 |
H11 |
110.162 |
| C2 |
O1 |
C3 |
112.989 |
|
C2 |
C4 |
C6 |
112.477 |
| C2 |
C4 |
H12 |
108.366 |
|
C2 |
C4 |
H13 |
108.366 |
| C3 |
C5 |
C7 |
112.477 |
|
C3 |
C5 |
H14 |
108.366 |
| C3 |
C5 |
H15 |
108.366 |
|
C4 |
C2 |
H8 |
110.314 |
| C4 |
C2 |
H9 |
110.314 |
|
C4 |
C6 |
H16 |
111.290 |
| C4 |
C6 |
H17 |
111.574 |
|
C4 |
C6 |
H18 |
111.574 |
| C5 |
C3 |
H10 |
110.314 |
|
C5 |
C3 |
H11 |
110.314 |
| C5 |
C7 |
H19 |
111.290 |
|
C5 |
C7 |
H20 |
111.574 |
| C5 |
C7 |
H21 |
111.574 |
|
C6 |
C4 |
H12 |
110.506 |
| C6 |
C4 |
H13 |
110.506 |
|
C7 |
C5 |
H14 |
110.506 |
| C7 |
C5 |
H15 |
110.506 |
|
H8 |
C2 |
H9 |
106.879 |
| H10 |
C3 |
H11 |
106.879 |
|
H12 |
C4 |
H13 |
106.399 |
| H14 |
C5 |
H15 |
106.399 |
|
H16 |
C6 |
H17 |
107.364 |
| H16 |
C6 |
H18 |
107.364 |
|
H17 |
C6 |
H18 |
107.442 |
| H19 |
C7 |
H20 |
107.364 |
|
H19 |
C7 |
H21 |
107.364 |
| H20 |
C7 |
H21 |
107.442 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.332 |
|
|
|
| 2 |
C |
-0.195 |
|
|
|
| 3 |
C |
-0.195 |
|
|
|
| 4 |
C |
-0.471 |
|
|
|
| 5 |
C |
-0.471 |
|
|
|
| 6 |
C |
-0.671 |
|
|
|
| 7 |
C |
-0.671 |
|
|
|
| 8 |
H |
0.189 |
|
|
|
| 9 |
H |
0.189 |
|
|
|
| 10 |
H |
0.189 |
|
|
|
| 11 |
H |
0.189 |
|
|
|
| 12 |
H |
0.226 |
|
|
|
| 13 |
H |
0.226 |
|
|
|
| 14 |
H |
0.226 |
|
|
|
| 15 |
H |
0.226 |
|
|
|
| 16 |
H |
0.231 |
|
|
|
| 17 |
H |
0.221 |
|
|
|
| 18 |
H |
0.221 |
|
|
|
| 19 |
H |
0.231 |
|
|
|
| 20 |
H |
0.221 |
|
|
|
| 21 |
H |
0.221 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.915 |
0.915 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.102 |
0.000 |
0.000 |
| y |
0.000 |
-44.221 |
0.000 |
| z |
0.000 |
0.000 |
-47.575 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.205 |
0.000 |
0.000 |
| y |
0.000 |
2.618 |
0.000 |
| z |
0.000 |
0.000 |
-2.413 |
|
| Polar |
|---|
| 3z2-r2 | -4.827 |
|---|
| x2-y2 | -1.882 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.572 |
0.000 |
0.000 |
| y |
0.000 |
13.543 |
0.000 |
| z |
0.000 |
0.000 |
10.036 |
<r2> (average value of r
2) Å
2
| <r2> |
431.330 |
| (<r2>)1/2 |
20.768 |
Jump to
S1C1
Energy calculated at HSEh1PBE/6-311G*
| | hartrees |
|---|
| Energy at 0K | -311.999288 |
| Energy at 298.15K | -312.015060 |
| HF Energy | -311.999288 |
| Nuclear repulsion energy | 326.225581 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3145 |
3018 |
8.88 |
|
|
|
| 2 |
A |
3120 |
2994 |
68.70 |
|
|
|
| 3 |
A |
3089 |
2965 |
12.60 |
|
|
|
| 4 |
A |
3054 |
2930 |
2.98 |
|
|
|
| 5 |
A |
3051 |
2928 |
9.12 |
|
|
|
| 6 |
A |
3004 |
2883 |
11.37 |
|
|
|
| 7 |
A |
2976 |
2856 |
106.18 |
|
|
|
| 8 |
A |
1544 |
1482 |
5.16 |
|
|
|
| 9 |
A |
1525 |
1463 |
5.45 |
|
|
|
| 10 |
A |
1510 |
1449 |
7.61 |
|
|
|
| 11 |
A |
1489 |
1429 |
0.93 |
|
|
|
| 12 |
A |
1463 |
1404 |
2.06 |
|
|
|
| 13 |
A |
1423 |
1365 |
3.22 |
|
|
|
| 14 |
A |
1386 |
1330 |
0.01 |
|
|
|
| 15 |
A |
1329 |
1276 |
3.72 |
|
|
|
| 16 |
A |
1284 |
1233 |
0.23 |
|
|
|
| 17 |
A |
1192 |
1144 |
7.22 |
|
|
|
| 18 |
A |
1156 |
1109 |
11.25 |
|
|
|
| 19 |
A |
1104 |
1060 |
8.34 |
|
|
|
| 20 |
A |
970 |
931 |
7.94 |
|
|
|
| 21 |
A |
929 |
892 |
7.41 |
|
|
|
| 22 |
A |
917 |
880 |
0.01 |
|
|
|
| 23 |
A |
757 |
726 |
0.17 |
|
|
|
| 24 |
A |
487 |
467 |
0.13 |
|
|
|
| 25 |
A |
337 |
323 |
0.30 |
|
|
|
| 26 |
A |
272 |
261 |
0.50 |
|
|
|
| 27 |
A |
168 |
161 |
0.05 |
|
|
|
| 28 |
A |
114 |
109 |
0.02 |
|
|
|
| 29 |
A |
46 |
44 |
0.00 |
|
|
|
| 30 |
B |
3144 |
3017 |
60.10 |
|
|
|
| 31 |
B |
3120 |
2994 |
36.50 |
|
|
|
| 32 |
B |
3089 |
2965 |
33.62 |
|
|
|
| 33 |
B |
3053 |
2930 |
80.85 |
|
|
|
| 34 |
B |
3051 |
2928 |
59.62 |
|
|
|
| 35 |
B |
3001 |
2880 |
111.19 |
|
|
|
| 36 |
B |
2964 |
2844 |
16.33 |
|
|
|
| 37 |
B |
1524 |
1463 |
1.25 |
|
|
|
| 38 |
B |
1523 |
1461 |
15.00 |
|
|
|
| 39 |
B |
1509 |
1448 |
13.38 |
|
|
|
| 40 |
B |
1489 |
1429 |
6.99 |
|
|
|
| 41 |
B |
1424 |
1366 |
19.08 |
|
|
|
| 42 |
B |
1410 |
1353 |
34.49 |
|
|
|
| 43 |
B |
1382 |
1326 |
11.10 |
|
|
|
| 44 |
B |
1320 |
1267 |
4.16 |
|
|
|
| 45 |
B |
1294 |
1241 |
6.99 |
|
|
|
| 46 |
B |
1197 |
1149 |
88.09 |
|
|
|
| 47 |
B |
1184 |
1136 |
106.03 |
|
|
|
| 48 |
B |
1122 |
1076 |
16.12 |
|
|
|
| 49 |
B |
1040 |
998 |
36.23 |
|
|
|
| 50 |
B |
942 |
904 |
1.20 |
|
|
|
| 51 |
B |
897 |
860 |
0.50 |
|
|
|
| 52 |
B |
790 |
758 |
2.79 |
|
|
|
| 53 |
B |
504 |
484 |
1.53 |
|
|
|
| 54 |
B |
327 |
314 |
0.45 |
|
|
|
| 55 |
B |
223 |
214 |
2.54 |
|
|
|
| 56 |
B |
171 |
165 |
4.33 |
|
|
|
| 57 |
B |
78 |
75 |
1.33 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42804.1 cm
-1
Scaled (by 0.9596) Zero Point Vibrational Energy (zpe) 41074.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-311G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.112 |
| C2 |
0.025 |
1.173 |
0.887 |
| C3 |
-0.025 |
-1.173 |
0.887 |
| C4 |
0.000 |
2.375 |
-0.033 |
| C5 |
0.000 |
-2.375 |
-0.033 |
| C6 |
-1.261 |
2.452 |
-0.879 |
| C7 |
1.261 |
-2.452 |
-0.879 |
| H8 |
-0.845 |
1.194 |
1.567 |
| H9 |
0.929 |
1.188 |
1.518 |
| H10 |
0.845 |
-1.194 |
1.567 |
| H11 |
-0.929 |
-1.188 |
1.518 |
| H12 |
0.885 |
2.335 |
-0.678 |
| H13 |
0.103 |
3.278 |
0.581 |
| H14 |
-0.885 |
-2.335 |
-0.678 |
| H15 |
-0.103 |
-3.278 |
0.581 |
| H16 |
-1.243 |
3.319 |
-1.545 |
| H17 |
-1.370 |
1.556 |
-1.493 |
| H18 |
-2.155 |
2.535 |
-0.252 |
| H19 |
1.243 |
-3.319 |
-1.545 |
| H20 |
1.370 |
-1.556 |
-1.493 |
| H21 |
2.155 |
-2.535 |
-0.252 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4064 | 1.4064 | 2.3798 | 2.3798 | 2.9304 | 2.9304 | 2.0625 | 2.0612 | 2.0625 | 2.0612 | 2.6189 | 3.3128 | 2.6189 | 3.3128 | 3.9127 | 2.6218 | 3.3469 | 3.9127 | 2.6218 | 3.3469 |
C2 | 1.4064 | | 2.3470 | 1.5139 | 3.6660 | 2.5320 | 4.2182 | 1.1037 | 1.1020 | 2.5954 | 2.6232 | 2.1305 | 2.1281 | 3.9477 | 4.4633 | 3.4828 | 2.7852 | 2.8111 | 5.2519 | 3.8630 | 4.4254 | C3 | 1.4064 | 2.3470 | | 3.6660 | 1.5139 | 4.2182 | 2.5320 | 2.5954 | 2.6232 | 1.1037 | 1.1020 | 3.9477 | 4.4633 | 2.1305 | 2.1281 | 5.2519 | 3.8630 | 4.4254 | 3.4828 | 2.7852 | 2.8111 | C4 | 2.3798 | 1.5139 | 3.6660 | | 4.7508 | 1.5211 | 5.0610 | 2.1604 | 2.1624 | 4.0012 | 3.9953 | 1.0958 | 1.0963 | 4.8358 | 5.6873 | 2.1736 | 2.1635 | 2.1718 | 6.0217 | 4.4119 | 5.3668 | C5 | 2.3798 | 3.6660 | 1.5139 | 4.7508 | | 5.0610 | 1.5211 | 4.0012 | 3.9953 | 2.1604 | 2.1624 | 4.8358 | 5.6873 | 1.0958 | 1.0963 | 6.0217 | 4.4119 | 5.3668 | 2.1736 | 2.1635 | 2.1718 | C6 | 2.9304 | 2.5320 | 4.2182 | 1.5211 | 5.0610 | | 5.5151 | 2.7820 | 3.4843 | 4.8694 | 4.3710 | 2.1585 | 2.1626 | 4.8060 | 6.0256 | 1.0932 | 1.0916 | 1.0951 | 6.3265 | 4.8337 | 6.0775 | C7 | 2.9304 | 4.2182 | 2.5320 | 5.0610 | 1.5211 | 5.5151 | | 4.8694 | 4.3710 | 2.7820 | 3.4843 | 4.8060 | 6.0256 | 2.1585 | 2.1626 | 6.3265 | 4.8337 | 6.0775 | 1.0932 | 1.0916 | 1.0951 | H8 | 2.0625 | 1.1037 | 2.5954 | 2.1604 | 4.0012 | 2.7820 | 4.8694 | | 1.7742 | 2.9248 | 2.3836 | 3.0547 | 2.4923 | 4.1823 | 4.6384 | 3.7893 | 3.1254 | 2.6117 | 5.8659 | 4.6719 | 5.1193 | H9 | 2.0612 | 1.1020 | 2.6232 | 2.1624 | 3.9953 | 3.4843 | 4.3710 | 1.7742 | | 2.3836 | 3.0157 | 2.4777 | 2.4343 | 4.5298 | 4.6779 | 4.3175 | 3.8056 | 3.8017 | 5.4582 | 4.0972 | 4.3004 | H10 | 2.0625 | 2.5954 | 1.1037 | 4.0012 | 2.1604 | 4.8694 | 2.7820 | 2.9248 | 2.3836 | | 1.7742 | 4.1823 | 4.6384 | 3.0547 | 2.4923 | 5.8659 | 4.6719 | 5.1193 | 3.7893 | 3.1254 | 2.6117 | H11 | 2.0612 | 2.6232 | 1.1020 | 3.9953 | 2.1624 | 4.3710 | 3.4843 | 2.3836 | 3.0157 | 1.7742 | | 4.5298 | 4.6779 | 2.4777 | 2.4343 | 5.4582 | 4.0972 | 4.3004 | 4.3175 | 3.8056 | 3.8017 | H12 | 2.6189 | 2.1305 | 3.9477 | 1.0958 | 4.8358 | 2.1585 | 4.8060 | 3.0547 | 2.4777 | 4.1823 | 4.5298 | | 1.7568 | 4.9935 | 5.8363 | 2.4997 | 2.5205 | 3.0756 | 5.7312 | 4.0047 | 5.0508 | H13 | 3.3128 | 2.1281 | 4.4633 | 1.0963 | 5.6873 | 2.1626 | 6.0256 | 2.4923 | 2.4343 | 4.6384 | 4.6779 | 1.7568 | | 5.8363 | 6.5587 | 2.5174 | 3.0720 | 2.5186 | 7.0243 | 5.4104 | 6.2203 | H14 | 2.6189 | 3.9477 | 2.1305 | 4.8358 | 1.0958 | 4.8060 | 2.1585 | 4.1823 | 4.5298 | 3.0547 | 2.4777 | 4.9935 | 5.8363 | | 1.7568 | 5.7312 | 4.0047 | 5.0508 | 2.4997 | 2.5205 | 3.0756 | H15 | 3.3128 | 4.4633 | 2.1281 | 5.6873 | 1.0963 | 6.0256 | 2.1626 | 4.6384 | 4.6779 | 2.4923 | 2.4343 | 5.8363 | 6.5587 | 1.7568 | | 7.0243 | 5.4104 | 6.2203 | 2.5174 | 3.0720 | 2.5186 | H16 | 3.9127 | 3.4828 | 5.2519 | 2.1736 | 6.0217 | 1.0932 | 6.3265 | 3.7893 | 4.3175 | 5.8659 | 5.4582 | 2.4997 | 2.5174 | 5.7312 | 7.0243 | | 1.7684 | 1.7660 | 7.0887 | 5.5315 | 6.8914 | H17 | 2.6218 | 2.7852 | 3.8630 | 2.1635 | 4.4119 | 1.0916 | 4.8337 | 3.1254 | 3.8056 | 4.6719 | 4.0972 | 2.5205 | 3.0720 | 4.0047 | 5.4104 | 1.7684 | | 1.7652 | 5.5315 | 4.1460 | 5.5407 | H18 | 3.3469 | 2.8111 | 4.4254 | 2.1718 | 5.3668 | 1.0951 | 6.0775 | 2.6117 | 3.8017 | 5.1193 | 4.3004 | 3.0756 | 2.5186 | 5.0508 | 6.2203 | 1.7660 | 1.7652 | | 6.8914 | 5.5407 | 6.6541 | H19 | 3.9127 | 5.2519 | 3.4828 | 6.0217 | 2.1736 | 6.3265 | 1.0932 | 5.8659 | 5.4582 | 3.7893 | 4.3175 | 5.7312 | 7.0243 | 2.4997 | 2.5174 | 7.0887 | 5.5315 | 6.8914 | | 1.7684 | 1.7660 | H20 | 2.6218 | 3.8630 | 2.7852 | 4.4119 | 2.1635 | 4.8337 | 1.0916 | 4.6719 | 4.0972 | 3.1254 | 3.8056 | 4.0047 | 5.4104 | 2.5205 | 3.0720 | 5.5315 | 4.1460 | 5.5407 | 1.7684 | | 1.7652 | H21 | 3.3469 | 4.4254 | 2.8111 | 5.3668 | 2.1718 | 6.0775 | 1.0951 | 5.1193 | 4.3004 | 2.6117 | 3.8017 | 5.0508 | 6.2203 | 3.0756 | 2.5186 | 6.8914 | 5.5407 | 6.6541 | 1.7660 | 1.7652 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
109.103 |
|
O1 |
C2 |
H8 |
109.926 |
| O1 |
C2 |
H9 |
109.927 |
|
O1 |
C3 |
C5 |
109.103 |
| O1 |
C3 |
H10 |
109.926 |
|
O1 |
C3 |
H11 |
109.927 |
| C2 |
O1 |
C3 |
113.110 |
|
C2 |
C4 |
C6 |
113.075 |
| C2 |
C4 |
H12 |
108.380 |
|
C2 |
C4 |
H13 |
108.163 |
| C3 |
C5 |
C7 |
113.075 |
|
C3 |
C5 |
H14 |
108.380 |
| C3 |
C5 |
H15 |
108.163 |
|
C4 |
C2 |
H8 |
110.258 |
| C4 |
C2 |
H9 |
110.515 |
|
C4 |
C6 |
H16 |
111.436 |
| C4 |
C6 |
H17 |
110.725 |
|
C4 |
C6 |
H18 |
111.178 |
| C5 |
C3 |
H10 |
110.258 |
|
C5 |
C3 |
H11 |
110.515 |
| C5 |
C7 |
H19 |
111.436 |
|
C5 |
C7 |
H20 |
110.725 |
| C5 |
C7 |
H21 |
111.178 |
|
C6 |
C4 |
H12 |
110.077 |
| C6 |
C4 |
H13 |
110.374 |
|
C7 |
C5 |
H14 |
110.077 |
| C7 |
C5 |
H15 |
110.374 |
|
H8 |
C2 |
H9 |
107.095 |
| H10 |
C3 |
H11 |
107.095 |
|
H12 |
C4 |
H13 |
106.533 |
| H14 |
C5 |
H15 |
106.533 |
|
H16 |
C6 |
H17 |
108.075 |
| H16 |
C6 |
H18 |
107.610 |
|
H17 |
C6 |
H18 |
107.655 |
| H19 |
C7 |
H20 |
108.075 |
|
H19 |
C7 |
H21 |
107.610 |
| H20 |
C7 |
H21 |
107.655 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.332 |
|
|
|
| 2 |
C |
-0.212 |
|
|
|
| 3 |
C |
-0.212 |
|
|
|
| 4 |
C |
-0.468 |
|
|
|
| 5 |
C |
-0.468 |
|
|
|
| 6 |
C |
-0.661 |
|
|
|
| 7 |
C |
-0.661 |
|
|
|
| 8 |
H |
0.190 |
|
|
|
| 9 |
H |
0.200 |
|
|
|
| 10 |
H |
0.190 |
|
|
|
| 11 |
H |
0.200 |
|
|
|
| 12 |
H |
0.226 |
|
|
|
| 13 |
H |
0.216 |
|
|
|
| 14 |
H |
0.226 |
|
|
|
| 15 |
H |
0.216 |
|
|
|
| 16 |
H |
0.222 |
|
|
|
| 17 |
H |
0.240 |
|
|
|
| 18 |
H |
0.214 |
|
|
|
| 19 |
H |
0.222 |
|
|
|
| 20 |
H |
0.240 |
|
|
|
| 21 |
H |
0.214 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.013 |
1.013 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.202 |
0.399 |
0.000 |
| y |
0.399 |
-44.231 |
0.000 |
| z |
0.000 |
0.000 |
-46.134 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.020 |
0.399 |
0.000 |
| y |
0.399 |
2.438 |
0.000 |
| z |
0.000 |
0.000 |
-0.418 |
|
| Polar |
|---|
| 3z2-r2 | -0.835 |
|---|
| x2-y2 | -2.972 |
|---|
| xy | 0.399 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.216 |
-0.488 |
0.000 |
| y |
-0.488 |
12.280 |
0.000 |
| z |
0.000 |
0.000 |
10.363 |
<r2> (average value of r
2) Å
2
| <r2> |
342.094 |
| (<r2>)1/2 |
18.496 |