Jump to
S1C2
Energy calculated at HSEh1PBE/6-31G
| | hartrees |
|---|
| Energy at 0K | -341.920666 |
| Energy at 298.15K | |
| HF Energy | -341.920666 |
| Nuclear repulsion energy | 241.616237 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3122 |
2987 |
33.30 |
|
|
|
| 2 |
A1 |
1889 |
1807 |
540.03 |
|
|
|
| 3 |
A1 |
1582 |
1514 |
0.15 |
|
|
|
| 4 |
A1 |
1400 |
1339 |
0.50 |
|
|
|
| 5 |
A1 |
1149 |
1099 |
190.29 |
|
|
|
| 6 |
A1 |
959 |
917 |
13.29 |
|
|
|
| 7 |
A1 |
794 |
759 |
37.26 |
|
|
|
| 8 |
A1 |
708 |
677 |
1.08 |
|
|
|
| 9 |
A2 |
3165 |
3027 |
0.00 |
|
|
|
| 10 |
A2 |
1267 |
1212 |
0.00 |
|
|
|
| 11 |
A2 |
1173 |
1123 |
0.00 |
|
|
|
| 12 |
A2 |
162i |
155i |
0.00 |
|
|
|
| 13 |
B1 |
3186 |
3048 |
32.53 |
|
|
|
| 14 |
B1 |
1265 |
1211 |
0.64 |
|
|
|
| 15 |
B1 |
883 |
845 |
0.17 |
|
|
|
| 16 |
B1 |
710 |
679 |
29.63 |
|
|
|
| 17 |
B1 |
145 |
139 |
4.95 |
|
|
|
| 18 |
B2 |
3116 |
2980 |
20.99 |
|
|
|
| 19 |
B2 |
1571 |
1503 |
3.80 |
|
|
|
| 20 |
B2 |
1421 |
1359 |
0.40 |
|
|
|
| 21 |
B2 |
1148 |
1099 |
5.24 |
|
|
|
| 22 |
B2 |
971 |
929 |
314.66 |
|
|
|
| 23 |
B2 |
746 |
714 |
3.42 |
|
|
|
| 24 |
B2 |
483 |
462 |
0.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16346.0 cm
-1
Scaled (by 0.9566) Zero Point Vibrational Energy (zpe) 15636.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-31G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.891 |
| O2 |
0.000 |
0.000 |
2.093 |
| O3 |
0.000 |
1.148 |
0.055 |
| O4 |
0.000 |
-1.148 |
0.055 |
| C5 |
0.000 |
0.777 |
-1.316 |
| C6 |
0.000 |
-0.777 |
-1.316 |
| H7 |
-0.890 |
1.192 |
-1.793 |
| H8 |
0.890 |
1.192 |
-1.793 |
| H9 |
0.890 |
-1.192 |
-1.793 |
| H10 |
-0.890 |
-1.192 |
-1.793 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.2024 | 1.4202 | 1.4202 | 2.3395 | 2.3395 | 3.0680 | 3.0680 | 3.0680 | 3.0680 |
O2 | 1.2024 | | 2.3394 | 2.3394 | 3.4965 | 3.4965 | 4.1607 | 4.1607 | 4.1607 | 4.1607 | O3 | 1.4202 | 2.3394 | | 2.2963 | 1.4202 | 2.3634 | 2.0511 | 2.0511 | 3.1114 | 3.1114 | O4 | 1.4202 | 2.3394 | 2.2963 | | 2.3634 | 1.4202 | 3.1114 | 3.1114 | 2.0511 | 2.0511 | C5 | 2.3395 | 3.4965 | 1.4202 | 2.3634 | | 1.5541 | 1.0914 | 1.0914 | 2.2126 | 2.2126 | C6 | 2.3395 | 3.4965 | 2.3634 | 1.4202 | 1.5541 | | 2.2126 | 2.2126 | 1.0914 | 1.0914 | H7 | 3.0680 | 4.1607 | 2.0511 | 3.1114 | 1.0914 | 2.2126 | | 1.7797 | 2.9748 | 2.3838 | H8 | 3.0680 | 4.1607 | 2.0511 | 3.1114 | 1.0914 | 2.2126 | 1.7797 | | 2.3838 | 2.9748 | H9 | 3.0680 | 4.1607 | 3.1114 | 2.0511 | 2.2126 | 1.0914 | 2.9748 | 2.3838 | | 1.7797 | H10 | 3.0680 | 4.1607 | 3.1114 | 2.0511 | 2.2126 | 1.0914 | 2.3838 | 2.9748 | 1.7797 | |
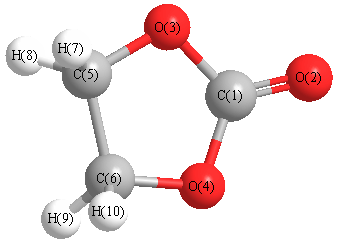 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.905 |
|
C1 |
O4 |
C6 |
110.905 |
| O2 |
C1 |
O3 |
126.054 |
|
O2 |
C1 |
O4 |
126.054 |
| O3 |
C1 |
O4 |
107.892 |
|
O3 |
C5 |
C6 |
105.149 |
| O3 |
C5 |
H7 |
108.798 |
|
O3 |
C5 |
H8 |
108.798 |
| O4 |
C6 |
C5 |
105.149 |
|
O4 |
C6 |
H9 |
108.798 |
| O4 |
C6 |
H10 |
108.798 |
|
C5 |
C6 |
H9 |
112.341 |
| C5 |
C6 |
H10 |
112.341 |
|
C6 |
C5 |
H7 |
112.341 |
| C6 |
C5 |
H8 |
112.341 |
|
H7 |
C5 |
H8 |
109.240 |
| H9 |
C6 |
H10 |
109.240 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.732 |
|
|
|
| 2 |
O |
-0.370 |
|
|
|
| 3 |
O |
-0.496 |
|
|
|
| 4 |
O |
-0.496 |
|
|
|
| 5 |
C |
-0.103 |
|
|
|
| 6 |
C |
-0.103 |
|
|
|
| 7 |
H |
0.209 |
|
|
|
| 8 |
H |
0.209 |
|
|
|
| 9 |
H |
0.209 |
|
|
|
| 10 |
H |
0.209 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.512 |
5.512 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.201 |
0.000 |
0.000 |
| y |
0.000 |
-37.802 |
0.000 |
| z |
0.000 |
0.000 |
-35.367 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.383 |
0.000 |
0.000 |
| y |
0.000 |
-4.018 |
0.000 |
| z |
0.000 |
0.000 |
-0.365 |
|
| Polar |
|---|
| 3z2-r2 | -0.731 |
|---|
| x2-y2 | 5.601 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.838 |
0.000 |
0.000 |
| y |
0.000 |
4.899 |
0.000 |
| z |
0.000 |
0.000 |
7.470 |
<r2> (average value of r
2) Å
2
| <r2> |
132.617 |
| (<r2>)1/2 |
11.516 |
Jump to
S1C1
Energy calculated at HSEh1PBE/6-31G
| | hartrees |
|---|
| Energy at 0K | -341.921165 |
| Energy at 298.15K | -341.927405 |
| HF Energy | -341.921165 |
| Nuclear repulsion energy | 242.046529 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3184 |
3046 |
6.78 |
|
|
|
| 2 |
A |
3106 |
2971 |
20.65 |
|
|
|
| 3 |
A |
1892 |
1810 |
525.64 |
|
|
|
| 4 |
A |
1570 |
1502 |
0.19 |
|
|
|
| 5 |
A |
1405 |
1344 |
0.70 |
|
|
|
| 6 |
A |
1267 |
1212 |
17.54 |
|
|
|
| 7 |
A |
1180 |
1129 |
20.58 |
|
|
|
| 8 |
A |
1131 |
1082 |
151.75 |
|
|
|
| 9 |
A |
965 |
923 |
9.49 |
|
|
|
| 10 |
A |
789 |
754 |
39.35 |
|
|
|
| 11 |
A |
705 |
674 |
1.39 |
|
|
|
| 12 |
A |
103 |
99 |
0.77 |
|
|
|
| 13 |
B |
3197 |
3058 |
21.63 |
|
|
|
| 14 |
B |
3108 |
2973 |
34.28 |
|
|
|
| 15 |
B |
1564 |
1496 |
5.14 |
|
|
|
| 16 |
B |
1418 |
1356 |
0.37 |
|
|
|
| 17 |
B |
1265 |
1210 |
1.71 |
|
|
|
| 18 |
B |
1132 |
1083 |
5.48 |
|
|
|
| 19 |
B |
974 |
932 |
253.82 |
|
|
|
| 20 |
B |
901 |
862 |
48.62 |
|
|
|
| 21 |
B |
722 |
690 |
27.96 |
|
|
|
| 22 |
B |
692 |
662 |
4.54 |
|
|
|
| 23 |
B |
484 |
463 |
0.51 |
|
|
|
| 24 |
B |
151 |
144 |
4.54 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16451.1 cm
-1
Scaled (by 0.9566) Zero Point Vibrational Energy (zpe) 15737.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-31G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.887 |
| O2 |
0.000 |
0.000 |
2.089 |
| O3 |
0.000 |
1.149 |
0.046 |
| O4 |
0.000 |
-1.149 |
0.046 |
| C5 |
0.184 |
0.749 |
-1.308 |
| C6 |
-0.184 |
-0.749 |
-1.308 |
| H7 |
-0.469 |
1.348 |
-1.942 |
| H8 |
1.226 |
0.916 |
-1.592 |
| H9 |
0.469 |
-1.348 |
-1.942 |
| H10 |
-1.226 |
-0.916 |
-1.592 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.2020 | 1.4236 | 1.4236 | 2.3259 | 2.3259 | 3.1681 | 2.9135 | 3.1681 | 2.9135 |
O2 | 1.2020 | | 2.3437 | 2.3437 | 3.4829 | 3.4829 | 4.2757 | 3.9866 | 4.2757 | 3.9866 | O3 | 1.4236 | 2.3437 | | 2.2976 | 1.4236 | 2.3380 | 2.0519 | 2.0594 | 3.2255 | 2.9073 | O4 | 1.4236 | 2.3437 | 2.2976 | | 2.3380 | 1.4236 | 3.2255 | 2.9073 | 2.0519 | 2.0594 | C5 | 2.3259 | 3.4829 | 1.4236 | 2.3380 | | 1.5415 | 1.0893 | 1.0938 | 2.2086 | 2.2002 | C6 | 2.3259 | 3.4829 | 2.3380 | 1.4236 | 1.5415 | | 2.2086 | 2.2002 | 1.0893 | 1.0938 | H7 | 3.1681 | 4.2757 | 2.0519 | 3.2255 | 1.0893 | 2.2086 | | 1.7837 | 2.8539 | 2.4131 | H8 | 2.9135 | 3.9866 | 2.0594 | 2.9073 | 1.0938 | 2.2002 | 1.7837 | | 2.4131 | 3.0619 | H9 | 3.1681 | 4.2757 | 3.2255 | 2.0519 | 2.2086 | 1.0893 | 2.8539 | 2.4131 | | 1.7837 | H10 | 2.9135 | 3.9866 | 2.9073 | 2.0594 | 2.2002 | 1.0938 | 2.4131 | 3.0619 | 1.7837 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
109.557 |
|
C1 |
O4 |
C6 |
109.557 |
| O2 |
C1 |
O3 |
126.199 |
|
O2 |
C1 |
O4 |
126.199 |
| O3 |
C1 |
O4 |
107.602 |
|
O3 |
C5 |
C6 |
104.024 |
| O3 |
C5 |
H7 |
108.759 |
|
O3 |
C5 |
H8 |
109.086 |
| O4 |
C6 |
C5 |
104.024 |
|
O4 |
C6 |
H9 |
108.759 |
| O4 |
C6 |
H10 |
109.086 |
|
C5 |
C6 |
H9 |
113.055 |
| C5 |
C6 |
H10 |
112.098 |
|
C6 |
C5 |
H7 |
113.055 |
| C6 |
C5 |
H8 |
112.098 |
|
H7 |
C5 |
H8 |
109.583 |
| H9 |
C6 |
H10 |
109.583 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.734 |
|
|
|
| 2 |
O |
-0.370 |
|
|
|
| 3 |
O |
-0.494 |
|
|
|
| 4 |
O |
-0.494 |
|
|
|
| 5 |
C |
-0.106 |
|
|
|
| 6 |
C |
-0.106 |
|
|
|
| 7 |
H |
0.211 |
|
|
|
| 8 |
H |
0.207 |
|
|
|
| 9 |
H |
0.211 |
|
|
|
| 10 |
H |
0.207 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.445 |
5.445 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.235 |
0.326 |
0.000 |
| y |
0.326 |
-37.841 |
0.000 |
| z |
0.000 |
0.000 |
-35.218 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.295 |
0.326 |
0.000 |
| y |
0.326 |
-4.115 |
0.000 |
| z |
0.000 |
0.000 |
-0.180 |
|
| Polar |
|---|
| 3z2-r2 | -0.361 |
|---|
| x2-y2 | 5.607 |
|---|
| xy | 0.326 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.907 |
0.110 |
0.000 |
| y |
0.110 |
4.857 |
0.000 |
| z |
0.000 |
0.000 |
7.451 |
<r2> (average value of r
2) Å
2
| <r2> |
131.716 |
| (<r2>)1/2 |
11.477 |