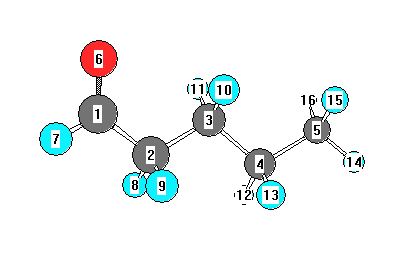
Vibrational Frequencies calculated at M06-2X/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3164 |
2997 |
30.00 |
|
|
|
| 2 |
A' |
3088 |
2924 |
35.41 |
|
|
|
| 3 |
A' |
3083 |
2919 |
13.72 |
|
|
|
| 4 |
A' |
3065 |
2902 |
19.95 |
|
|
|
| 5 |
A' |
3058 |
2896 |
16.65 |
|
|
|
| 6 |
A' |
2981 |
2823 |
138.13 |
|
|
|
| 7 |
A' |
1891 |
1790 |
145.29 |
|
|
|
| 8 |
A' |
1538 |
1457 |
5.66 |
|
|
|
| 9 |
A' |
1523 |
1442 |
0.98 |
|
|
|
| 10 |
A' |
1513 |
1433 |
1.45 |
|
|
|
| 11 |
A' |
1485 |
1407 |
12.99 |
|
|
|
| 12 |
A' |
1446 |
1369 |
7.28 |
|
|
|
| 13 |
A' |
1441 |
1365 |
6.24 |
|
|
|
| 14 |
A' |
1434 |
1358 |
8.64 |
|
|
|
| 15 |
A' |
1389 |
1315 |
13.89 |
|
|
|
| 16 |
A' |
1287 |
1219 |
3.44 |
|
|
|
| 17 |
A' |
1155 |
1094 |
10.93 |
|
|
|
| 18 |
A' |
1097 |
1039 |
0.89 |
|
|
|
| 19 |
A' |
1064 |
1008 |
1.13 |
|
|
|
| 20 |
A' |
940 |
890 |
1.43 |
|
|
|
| 21 |
A' |
917 |
868 |
11.02 |
|
|
|
| 22 |
A' |
699 |
662 |
14.67 |
|
|
|
| 23 |
A' |
402 |
380 |
1.75 |
|
|
|
| 24 |
A' |
299 |
283 |
4.91 |
|
|
|
| 25 |
A' |
141 |
133 |
5.16 |
|
|
|
| 26 |
A" |
3156 |
2989 |
46.70 |
|
|
|
| 27 |
A" |
3132 |
2966 |
24.26 |
|
|
|
| 28 |
A" |
3098 |
2934 |
5.24 |
|
|
|
| 29 |
A" |
3089 |
2925 |
7.50 |
|
|
|
| 30 |
A" |
1534 |
1452 |
7.28 |
|
|
|
| 31 |
A" |
1337 |
1266 |
0.26 |
|
|
|
| 32 |
A" |
1324 |
1253 |
0.07 |
|
|
|
| 33 |
A" |
1250 |
1184 |
0.01 |
|
|
|
| 34 |
A" |
1174 |
1112 |
0.03 |
|
|
|
| 35 |
A" |
988 |
935 |
0.98 |
|
|
|
| 36 |
A" |
867 |
821 |
0.11 |
|
|
|
| 37 |
A" |
751 |
711 |
1.53 |
|
|
|
| 38 |
A" |
685 |
649 |
3.26 |
|
|
|
| 39 |
A" |
259 |
245 |
0.02 |
|
|
|
| 40 |
A" |
196 |
185 |
1.43 |
|
|
|
| 41 |
A" |
116 |
110 |
0.67 |
|
|
|
| 42 |
A" |
81 |
76 |
2.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31566.0 cm
-1
Scaled (by 0.947) Zero Point Vibrational Energy (zpe) 29893.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.252 |
|
|
|
| 2 |
C |
-0.399 |
|
|
|
| 3 |
C |
-0.280 |
|
|
|
| 4 |
C |
-0.294 |
|
|
|
| 5 |
C |
-0.479 |
|
|
|
| 6 |
O |
-0.394 |
|
|
|
| 7 |
H |
0.127 |
|
|
|
| 8 |
H |
0.179 |
|
|
|
| 9 |
H |
0.179 |
|
|
|
| 10 |
H |
0.167 |
|
|
|
| 11 |
H |
0.167 |
|
|
|
| 12 |
H |
0.148 |
|
|
|
| 13 |
H |
0.148 |
|
|
|
| 14 |
H |
0.161 |
|
|
|
| 15 |
H |
0.160 |
|
|
|
| 16 |
H |
0.160 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.530 |
-0.195 |
0.000 |
2.537 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.488 |
3.818 |
0.000 |
| y |
3.818 |
-36.353 |
0.000 |
| z |
0.000 |
0.000 |
-37.123 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.750 |
3.818 |
0.000 |
| y |
3.818 |
4.452 |
0.000 |
| z |
0.000 |
0.000 |
3.298 |
|
| Polar |
|---|
| 3z2-r2 | 6.595 |
|---|
| x2-y2 | -8.135 |
|---|
| xy | 3.818 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.424 |
-0.385 |
0.000 |
| y |
-0.385 |
9.247 |
0.000 |
| z |
0.000 |
0.000 |
6.648 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |