Jump to
S1C2
S1C3
Vibrational Frequencies calculated at M06-2X/6-31G*
Geometric Data calculated at M06-2X/6-31G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Vibrational Frequencies calculated at M06-2X/6-31G*
Geometric Data calculated at M06-2X/6-31G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at M06-2X/6-31G*
| | hartrees |
|---|
| Energy at 0K | -345.635923 |
| Energy at 298.15K | |
| HF Energy | -345.635923 |
| Nuclear repulsion energy | 297.737015 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3213 |
3043 |
4.83 |
|
|
|
| 2 |
A |
3152 |
2985 |
2.59 |
|
|
|
| 3 |
A |
3126 |
2960 |
1.06 |
|
|
|
| 4 |
A |
3082 |
2918 |
0.42 |
|
|
|
| 5 |
A |
1891 |
1791 |
21.71 |
|
|
|
| 6 |
A |
1504 |
1424 |
16.17 |
|
|
|
| 7 |
A |
1502 |
1422 |
6.83 |
|
|
|
| 8 |
A |
1487 |
1408 |
10.03 |
|
|
|
| 9 |
A |
1415 |
1340 |
10.27 |
|
|
|
| 10 |
A |
1296 |
1228 |
26.72 |
|
|
|
| 11 |
A |
1160 |
1098 |
1.35 |
|
|
|
| 12 |
A |
1091 |
1033 |
0.53 |
|
|
|
| 13 |
A |
958 |
907 |
0.57 |
|
|
|
| 14 |
A |
811 |
768 |
0.17 |
|
|
|
| 15 |
A |
631 |
597 |
1.32 |
|
|
|
| 16 |
A |
497 |
471 |
7.83 |
|
|
|
| 17 |
A |
323 |
306 |
0.83 |
|
|
|
| 18 |
A |
180 |
171 |
0.17 |
|
|
|
| 19 |
A |
137 |
130 |
0.93 |
|
|
|
| 20 |
A |
65 |
62 |
7.11 |
|
|
|
| 21 |
B |
3213 |
3042 |
4.08 |
|
|
|
| 22 |
B |
3198 |
3028 |
3.42 |
|
|
|
| 23 |
B |
3152 |
2985 |
0.33 |
|
|
|
| 24 |
B |
3082 |
2918 |
1.41 |
|
|
|
| 25 |
B |
1867 |
1768 |
317.88 |
|
|
|
| 26 |
B |
1502 |
1422 |
2.02 |
|
|
|
| 27 |
B |
1497 |
1418 |
22.91 |
|
|
|
| 28 |
B |
1415 |
1340 |
103.26 |
|
|
|
| 29 |
B |
1280 |
1212 |
145.84 |
|
|
|
| 30 |
B |
1213 |
1149 |
117.84 |
|
|
|
| 31 |
B |
1075 |
1018 |
2.94 |
|
|
|
| 32 |
B |
1010 |
957 |
0.84 |
|
|
|
| 33 |
B |
914 |
865 |
11.31 |
|
|
|
| 34 |
B |
823 |
779 |
7.17 |
|
|
|
| 35 |
B |
553 |
523 |
26.93 |
|
|
|
| 36 |
B |
500 |
473 |
1.33 |
|
|
|
| 37 |
B |
418 |
395 |
1.27 |
|
|
|
| 38 |
B |
181 |
171 |
0.50 |
|
|
|
| 39 |
B |
29 |
28 |
11.70 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 27219.4 cm
-1
Scaled (by 0.947) Zero Point Vibrational Energy (zpe) 25776.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-31G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.035 |
| C2 |
0.000 |
1.219 |
0.111 |
| C3 |
0.000 |
-1.219 |
0.111 |
| C4 |
-1.358 |
1.716 |
-0.318 |
| C5 |
1.358 |
-1.716 |
-0.318 |
| O6 |
1.040 |
1.691 |
-0.286 |
| O7 |
-1.040 |
-1.691 |
-0.286 |
| H8 |
-0.902 |
-0.030 |
1.650 |
| H9 |
0.902 |
0.030 |
1.650 |
| H10 |
-1.252 |
2.462 |
-1.105 |
| H11 |
-1.962 |
0.869 |
-0.662 |
| H12 |
-1.877 |
2.154 |
0.542 |
| H13 |
1.252 |
-2.462 |
-1.105 |
| H14 |
1.962 |
-0.869 |
-0.662 |
| H15 |
1.877 |
-2.154 |
0.542 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
O7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
| C1 | | 1.5295 | 1.5295 | 2.5722 | 2.5722 | 2.3843 | 2.3843 | 1.0922 | 1.0922 | 3.4943 | 2.7357 | 2.8993 | 3.4943 | 2.7357 | 2.8993 |
C2 | 1.5295 | | 2.4381 | 1.5079 | 3.2618 | 1.2092 | 3.1153 | 2.1775 | 2.1441 | 2.1430 | 2.1375 | 2.1405 | 4.0742 | 2.9676 | 3.8841 | C3 | 1.5295 | 2.4381 | | 3.2618 | 1.5079 | 3.1153 | 1.2092 | 2.1441 | 2.1775 | 4.0742 | 2.9676 | 3.8841 | 2.1430 | 2.1375 | 2.1405 | C4 | 2.5722 | 1.5079 | 3.2618 | | 4.3757 | 2.3982 | 3.4211 | 2.6696 | 3.4382 | 1.0901 | 1.0956 | 1.0956 | 4.9887 | 4.2211 | 5.1162 | C5 | 2.5722 | 3.2618 | 1.5079 | 4.3757 | | 3.4211 | 2.3982 | 3.4382 | 2.6696 | 4.9887 | 4.2211 | 5.1162 | 1.0901 | 1.0956 | 1.0956 | O6 | 2.3843 | 1.2092 | 3.1153 | 2.3982 | 3.4211 | | 3.9697 | 3.2373 | 2.5550 | 2.5536 | 3.1348 | 3.0672 | 4.2380 | 2.7462 | 4.0209 | O7 | 2.3843 | 3.1153 | 1.2092 | 3.4211 | 2.3982 | 3.9697 | | 2.5550 | 3.2373 | 4.2380 | 2.7462 | 4.0209 | 2.5536 | 3.1348 | 3.0672 | H8 | 1.0922 | 2.1775 | 2.1441 | 2.6696 | 3.4382 | 3.2373 | 2.5550 | | 1.8045 | 3.7314 | 2.6981 | 2.6360 | 4.2602 | 3.7753 | 3.6692 | H9 | 1.0922 | 2.1441 | 2.1775 | 3.4382 | 2.6696 | 2.5550 | 3.2373 | 1.8045 | | 4.2602 | 3.7753 | 3.6692 | 3.7314 | 2.6981 | 2.6360 | H10 | 3.4943 | 2.1430 | 4.0742 | 1.0901 | 4.9887 | 2.5536 | 4.2380 | 3.7314 | 4.2602 | | 1.7993 | 1.7877 | 5.5249 | 4.6500 | 5.8150 | H11 | 2.7357 | 2.1375 | 2.9676 | 1.0956 | 4.2211 | 3.1348 | 2.7462 | 2.6981 | 3.7753 | 1.7993 | | 1.7633 | 4.6500 | 4.2910 | 5.0321 | H12 | 2.8993 | 2.1405 | 3.8841 | 1.0956 | 5.1162 | 3.0672 | 4.0209 | 2.6360 | 3.6692 | 1.7877 | 1.7633 | | 5.8150 | 5.0321 | 5.7140 | H13 | 3.4943 | 4.0742 | 2.1430 | 4.9887 | 1.0901 | 4.2380 | 2.5536 | 4.2602 | 3.7314 | 5.5249 | 4.6500 | 5.8150 | | 1.7993 | 1.7877 | H14 | 2.7357 | 2.9676 | 2.1375 | 4.2211 | 1.0956 | 2.7462 | 3.1348 | 3.7753 | 2.6981 | 4.6500 | 4.2910 | 5.0321 | 1.7993 | | 1.7633 | H15 | 2.8993 | 3.8841 | 2.1405 | 5.1162 | 1.0956 | 4.0209 | 3.0672 | 3.6692 | 2.6360 | 5.8150 | 5.0321 | 5.7140 | 1.7877 | 1.7633 | |
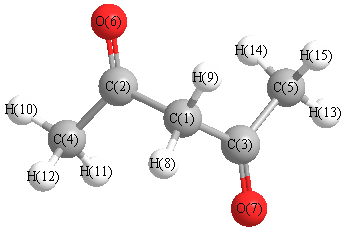 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
115.738 |
|
C1 |
C2 |
O6 |
120.614 |
| C1 |
C3 |
C5 |
115.738 |
|
C1 |
C3 |
O7 |
120.614 |
| C2 |
C1 |
C3 |
105.692 |
|
C2 |
C1 |
H8 |
111.218 |
| C2 |
C1 |
H9 |
108.587 |
|
C2 |
C4 |
H10 |
110.113 |
| C2 |
C4 |
H11 |
109.349 |
|
C2 |
C4 |
H12 |
109.582 |
| C3 |
C1 |
H8 |
108.587 |
|
C3 |
C1 |
H9 |
111.218 |
| C3 |
C5 |
H13 |
110.113 |
|
C3 |
C5 |
H14 |
109.349 |
| C3 |
C5 |
H15 |
109.582 |
|
C4 |
C2 |
O6 |
123.547 |
| C5 |
C3 |
O7 |
123.547 |
|
H8 |
C1 |
H9 |
111.403 |
| H10 |
C4 |
H11 |
110.820 |
|
H10 |
C4 |
H12 |
109.758 |
| H11 |
C4 |
H12 |
107.166 |
|
H13 |
C5 |
H14 |
110.820 |
| H13 |
C5 |
H15 |
109.758 |
|
H14 |
C5 |
H15 |
107.166 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.452 |
|
|
|
| 2 |
C |
0.424 |
|
|
|
| 3 |
C |
0.424 |
|
|
|
| 4 |
C |
-0.566 |
|
|
|
| 5 |
C |
-0.566 |
|
|
|
| 6 |
O |
-0.429 |
|
|
|
| 7 |
O |
-0.429 |
|
|
|
| 8 |
H |
0.197 |
|
|
|
| 9 |
H |
0.197 |
|
|
|
| 10 |
H |
0.200 |
|
|
|
| 11 |
H |
0.210 |
|
|
|
| 12 |
H |
0.190 |
|
|
|
| 13 |
H |
0.200 |
|
|
|
| 14 |
H |
0.210 |
|
|
|
| 15 |
H |
0.190 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.522 |
1.522 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-43.344 |
-8.939 |
0.000 |
| y |
-8.939 |
-45.219 |
0.000 |
| z |
0.000 |
0.000 |
-39.959 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.754 |
-8.939 |
0.000 |
| y |
-8.939 |
-3.568 |
0.000 |
| z |
0.000 |
0.000 |
4.322 |
|
| Polar |
|---|
| 3z2-r2 | 8.644 |
|---|
| x2-y2 | 1.876 |
|---|
| xy | -8.939 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.905 |
-0.163 |
0.000 |
| y |
-0.163 |
8.671 |
0.000 |
| z |
0.000 |
0.000 |
6.810 |
<r2> (average value of r
2) Å
2
| <r2> |
225.939 |
| (<r2>)1/2 |
15.031 |