Jump to
S1C2
Energy calculated at M06-2X/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -2611.161088 |
| Energy at 298.15K | -2611.164989 |
| HF Energy | -2611.161088 |
| Nuclear repulsion energy | 81.557227 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3215 |
3062 |
4.24 |
|
|
|
| 2 |
A' |
1410 |
1343 |
15.63 |
|
|
|
| 3 |
A' |
772 |
736 |
18.10 |
|
|
|
| 4 |
A' |
323 |
307 |
50.99 |
|
|
|
| 5 |
A" |
3365 |
3204 |
0.45 |
|
|
|
| 6 |
A" |
955 |
909 |
0.57 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5019.6 cm
-1
Scaled (by 0.9524) Zero Point Vibrational Energy (zpe) 4780.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-31G(2df,p)
Point Group is Cs
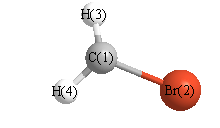
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.006 |
1.467 |
0.000 |
| Br2 |
-0.006 |
-0.364 |
0.000 |
| H3 |
0.114 |
1.970 |
0.945 |
| H4 |
0.114 |
1.970 |
-0.945 |
Atom - Atom Distances (Å)
| |
C1 |
Br2 |
H3 |
H4 |
| C1 | | 1.8309 | 1.0778 | 1.0778 |
Br2 | 1.8309 | | 2.5215 | 2.5215 | H3 | 1.0778 | 2.5215 | | 1.8906 | H4 | 1.0778 | 2.5215 | 1.8906 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Br2 |
C1 |
H3 |
117.856 |
|
Br2 |
C1 |
H4 |
117.856 |
| H3 |
C1 |
H4 |
122.589 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.397 |
|
|
|
| 2 |
Br |
0.039 |
|
|
|
| 3 |
H |
0.179 |
|
|
|
| 4 |
H |
0.179 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.184 |
1.100 |
0.000 |
1.115 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.905 |
0.419 |
0.000 |
| y |
0.419 |
-21.615 |
0.000 |
| z |
0.000 |
0.000 |
-24.104 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.045 |
0.419 |
0.000 |
| y |
0.419 |
3.389 |
0.000 |
| z |
0.000 |
0.000 |
-0.344 |
|
| Polar |
|---|
| 3z2-r2 | -0.689 |
|---|
| x2-y2 | -4.290 |
|---|
| xy | 0.419 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.181 |
0.043 |
0.000 |
| y |
0.043 |
5.757 |
0.000 |
| z |
0.000 |
0.000 |
3.821 |
<r2> (average value of r
2) Å
2
| <r2> |
42.041 |
| (<r2>)1/2 |
6.484 |
Jump to
S1C1
Energy calculated at M06-2X/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -2611.161034 |
| Energy at 298.15K | |
| HF Energy | -2611.161034 |
| Nuclear repulsion energy | 81.615029 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3218 |
3064 |
3.58 |
107.40 |
0.11 |
0.21 |
| 2 |
A1 |
1405 |
1338 |
16.12 |
2.92 |
0.74 |
0.85 |
| 3 |
A1 |
762 |
725 |
18.15 |
7.59 |
0.12 |
0.21 |
| 4 |
B1 |
166i |
158i |
55.86 |
0.29 |
0.75 |
0.86 |
| 5 |
B2 |
3370 |
3210 |
0.71 |
58.51 |
0.75 |
0.86 |
| 6 |
B2 |
949 |
904 |
0.51 |
2.14 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4768.5 cm
-1
Scaled (by 0.9524) Zero Point Vibrational Energy (zpe) 4541.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-31G(2df,p)
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.465 |
| Br2 |
0.000 |
0.000 |
0.364 |
| H3 |
0.000 |
0.948 |
-1.976 |
| H4 |
0.000 |
-0.948 |
-1.976 |
Atom - Atom Distances (Å)
| |
C1 |
Br2 |
H3 |
H4 |
| C1 | | 1.8287 | 1.0770 | 1.0770 |
Br2 | 1.8287 | | 2.5247 | 2.5247 | H3 | 1.0770 | 2.5247 | | 1.8957 | H4 | 1.0770 | 2.5247 | 1.8957 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Br2 |
C1 |
H3 |
118.343 |
|
Br2 |
C1 |
H4 |
118.343 |
| H3 |
C1 |
H4 |
123.315 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.399 |
|
|
|
| 2 |
Br |
0.040 |
|
|
|
| 3 |
H |
0.179 |
|
|
|
| 4 |
H |
0.179 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.103 |
1.103 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.937 |
0.000 |
0.000 |
| y |
0.000 |
-24.077 |
0.000 |
| z |
0.000 |
0.000 |
-21.567 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.115 |
0.000 |
0.000 |
| y |
0.000 |
-0.325 |
0.000 |
| z |
0.000 |
0.000 |
3.440 |
|
| Polar |
|---|
| 3z2-r2 | 6.880 |
|---|
| x2-y2 | -1.860 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.204 |
0.000 |
0.000 |
| y |
0.000 |
3.849 |
0.000 |
| z |
0.000 |
0.000 |
5.795 |
<r2> (average value of r
2) Å
2
| <r2> |
42.019 |
| (<r2>)1/2 |
6.482 |