Jump to
S1C2
Energy calculated at M06-2X/3-21G*
| | hartrees |
|---|
| Energy at 0K | -155.052301 |
| Energy at 298.15K | -155.057733 |
| HF Energy | -155.052301 |
| Nuclear repulsion energy | 103.697842 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3236 |
3065 |
0.00 |
|
|
|
| 2 |
Ag |
3154 |
2988 |
0.00 |
|
|
|
| 3 |
Ag |
3135 |
2970 |
0.00 |
|
|
|
| 4 |
Ag |
1743 |
1651 |
0.00 |
|
|
|
| 5 |
Ag |
1526 |
1446 |
0.00 |
|
|
|
| 6 |
Ag |
1361 |
1290 |
0.00 |
|
|
|
| 7 |
Ag |
1256 |
1189 |
0.00 |
|
|
|
| 8 |
Ag |
909 |
861 |
0.00 |
|
|
|
| 9 |
Ag |
541 |
512 |
0.00 |
|
|
|
| 10 |
Au |
1090 |
1032 |
38.21 |
|
|
|
| 11 |
Au |
1003 |
950 |
120.02 |
|
|
|
| 12 |
Au |
548 |
519 |
17.58 |
|
|
|
| 13 |
Au |
149 |
142 |
0.47 |
|
|
|
| 14 |
Bg |
1049 |
994 |
0.00 |
|
|
|
| 15 |
Bg |
1001 |
948 |
0.00 |
|
|
|
| 16 |
Bg |
798 |
756 |
0.00 |
|
|
|
| 17 |
Bu |
3236 |
3066 |
13.44 |
|
|
|
| 18 |
Bu |
3154 |
2988 |
1.85 |
|
|
|
| 19 |
Bu |
3147 |
2981 |
10.89 |
|
|
|
| 20 |
Bu |
1694 |
1604 |
11.29 |
|
|
|
| 21 |
Bu |
1474 |
1396 |
5.79 |
|
|
|
| 22 |
Bu |
1371 |
1299 |
1.80 |
|
|
|
| 23 |
Bu |
1053 |
998 |
5.33 |
|
|
|
| 24 |
Bu |
310 |
294 |
1.59 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18970.3 cm
-1
Scaled (by 0.9472) Zero Point Vibrational Energy (zpe) 17968.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/3-21G*
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.330 |
0.656 |
0.000 |
| C2 |
0.330 |
-0.656 |
0.000 |
| C3 |
0.330 |
1.816 |
0.000 |
| C4 |
-0.330 |
-1.816 |
0.000 |
| H5 |
-1.418 |
0.645 |
0.000 |
| H6 |
1.418 |
-0.645 |
0.000 |
| H7 |
-0.187 |
2.768 |
0.000 |
| H8 |
1.415 |
1.848 |
0.000 |
| H9 |
0.187 |
-2.768 |
0.000 |
| H10 |
-1.415 |
-1.848 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4681 | 1.3350 | 2.4717 | 1.0878 | 2.1788 | 2.1173 | 2.1138 | 3.4626 | 2.7286 |
C2 | 1.4681 | | 2.4717 | 1.3350 | 2.1788 | 1.0878 | 3.4626 | 2.7286 | 2.1173 | 2.1138 | C3 | 1.3350 | 2.4717 | | 3.6916 | 2.1039 | 2.6908 | 1.0833 | 1.0858 | 4.5864 | 4.0584 | C4 | 2.4717 | 1.3350 | 3.6916 | | 2.6908 | 2.1039 | 4.5864 | 4.0584 | 1.0833 | 1.0858 | H5 | 1.0878 | 2.1788 | 2.1039 | 2.6908 | | 3.1154 | 2.4541 | 3.0779 | 3.7716 | 2.4930 | H6 | 2.1788 | 1.0878 | 2.6908 | 2.1039 | 3.1154 | | 3.7716 | 2.4930 | 2.4541 | 3.0779 | H7 | 2.1173 | 3.4626 | 1.0833 | 4.5864 | 2.4541 | 3.7716 | | 1.8477 | 5.5489 | 4.7767 | H8 | 2.1138 | 2.7286 | 1.0858 | 4.0584 | 3.0779 | 2.4930 | 1.8477 | | 4.7767 | 4.6553 | H9 | 3.4626 | 2.1173 | 4.5864 | 1.0833 | 3.7716 | 2.4541 | 5.5489 | 4.7767 | | 1.8477 | H10 | 2.7286 | 2.1138 | 4.0584 | 1.0858 | 2.4930 | 3.0779 | 4.7767 | 4.6553 | 1.8477 | |
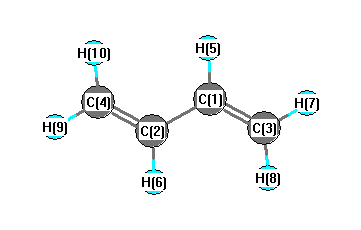 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
123.640 |
|
C1 |
C2 |
H6 |
116.168 |
| C1 |
C3 |
H7 |
121.868 |
|
C1 |
C3 |
H8 |
121.316 |
| C2 |
C1 |
C3 |
123.640 |
|
C2 |
C1 |
H5 |
116.168 |
| C2 |
C4 |
H9 |
121.868 |
|
C2 |
C4 |
H10 |
121.316 |
| C3 |
C1 |
H5 |
120.192 |
|
C4 |
C2 |
H6 |
120.192 |
| H7 |
C3 |
H8 |
116.817 |
|
H9 |
C4 |
H10 |
116.817 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.218 |
|
|
|
| 2 |
C |
-0.218 |
|
|
|
| 3 |
C |
-0.387 |
|
|
|
| 4 |
C |
-0.387 |
|
|
|
| 5 |
H |
0.205 |
|
|
|
| 6 |
H |
0.205 |
|
|
|
| 7 |
H |
0.203 |
|
|
|
| 8 |
H |
0.197 |
|
|
|
| 9 |
H |
0.203 |
|
|
|
| 10 |
H |
0.197 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-22.514 |
0.143 |
0.000 |
| y |
0.143 |
-22.455 |
0.000 |
| z |
0.000 |
0.000 |
-28.914 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.170 |
0.143 |
0.000 |
| y |
0.143 |
3.259 |
0.000 |
| z |
0.000 |
0.000 |
-6.429 |
|
| Polar |
|---|
| 3z2-r2 | -12.858 |
|---|
| x2-y2 | -0.060 |
|---|
| xy | 0.143 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.682 |
1.558 |
0.000 |
| y |
1.558 |
10.188 |
0.000 |
| z |
0.000 |
0.000 |
1.669 |
<r2> (average value of r
2) Å
2
| <r2> |
93.816 |
| (<r2>)1/2 |
9.686 |
Jump to
S1C1
Energy calculated at M06-2X/3-21G*
| | hartrees |
|---|
| Energy at 0K | -155.048642 |
| Energy at 298.15K | -155.054145 |
| HF Energy | -155.048642 |
| Nuclear repulsion energy | 105.003590 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3271 |
3098 |
4.01 |
|
|
|
| 2 |
A |
3195 |
3026 |
1.64 |
|
|
|
| 3 |
A |
3180 |
3013 |
7.93 |
|
|
|
| 4 |
A |
1713 |
1623 |
1.59 |
|
|
|
| 5 |
A |
1520 |
1440 |
11.09 |
|
|
|
| 6 |
A |
1387 |
1314 |
0.14 |
|
|
|
| 7 |
A |
1110 |
1051 |
0.15 |
|
|
|
| 8 |
A |
1053 |
997 |
0.64 |
|
|
|
| 9 |
A |
1017 |
963 |
9.40 |
|
|
|
| 10 |
A |
898 |
850 |
0.34 |
|
|
|
| 11 |
A |
784 |
742 |
5.99 |
|
|
|
| 12 |
A |
300 |
284 |
0.02 |
|
|
|
| 13 |
A |
188 |
178 |
0.05 |
|
|
|
| 14 |
B |
3269 |
3096 |
10.33 |
|
|
|
| 15 |
B |
3188 |
3020 |
5.11 |
|
|
|
| 16 |
B |
3174 |
3006 |
4.68 |
|
|
|
| 17 |
B |
1736 |
1645 |
1.73 |
|
|
|
| 18 |
B |
1500 |
1421 |
2.39 |
|
|
|
| 19 |
B |
1356 |
1285 |
0.27 |
|
|
|
| 20 |
B |
1153 |
1093 |
8.22 |
|
|
|
| 21 |
B |
1065 |
1008 |
29.43 |
|
|
|
| 22 |
B |
1018 |
965 |
104.57 |
|
|
|
| 23 |
B |
654 |
619 |
8.00 |
|
|
|
| 24 |
B |
494 |
467 |
17.48 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19110.6 cm
-1
Scaled (by 0.9472) Zero Point Vibrational Energy (zpe) 18101.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/3-21G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.112 |
0.731 |
0.558 |
| C2 |
-0.112 |
-0.731 |
0.558 |
| C3 |
-0.112 |
1.519 |
-0.494 |
| C4 |
0.112 |
-1.519 |
-0.494 |
| H5 |
0.468 |
1.162 |
1.491 |
| H6 |
-0.468 |
-1.162 |
1.491 |
| H7 |
0.086 |
2.585 |
-0.461 |
| H8 |
-0.517 |
1.122 |
-1.419 |
| H9 |
-0.086 |
-2.585 |
-0.461 |
| H10 |
0.517 |
-1.122 |
-1.419 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4789 | 1.3337 | 2.4842 | 1.0877 | 2.1889 | 2.1156 | 2.1120 | 3.4743 | 2.7404 |
C2 | 1.4789 | | 2.4842 | 1.3337 | 2.1889 | 1.0877 | 3.4743 | 2.7404 | 2.1156 | 2.1120 | C3 | 1.3337 | 2.4842 | | 3.0472 | 2.0985 | 3.3554 | 1.0837 | 1.0859 | 4.1042 | 2.8690 | C4 | 2.4842 | 1.3337 | 3.0472 | | 3.3554 | 2.0985 | 4.1042 | 2.8690 | 1.0837 | 1.0859 | H5 | 1.0877 | 2.1889 | 2.0985 | 3.3554 | | 2.5063 | 2.4454 | 3.0732 | 4.2611 | 3.7004 | H6 | 2.1889 | 1.0877 | 3.3554 | 2.0985 | 2.5063 | | 4.2611 | 3.7004 | 2.4454 | 3.0732 | H7 | 2.1156 | 3.4743 | 1.0837 | 4.1042 | 2.4454 | 4.2611 | | 1.8495 | 5.1719 | 3.8528 | H8 | 2.1120 | 2.7404 | 1.0859 | 2.8690 | 3.0732 | 3.7004 | 1.8495 | | 3.8528 | 2.4712 | H9 | 3.4743 | 2.1156 | 4.1042 | 1.0837 | 4.2611 | 2.4454 | 5.1719 | 3.8528 | | 1.8495 | H10 | 2.7404 | 2.1120 | 2.8690 | 1.0859 | 3.7004 | 3.0732 | 3.8528 | 2.4712 | 1.8495 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
123.990 |
|
C1 |
C2 |
H6 |
116.208 |
| C1 |
C3 |
H7 |
121.782 |
|
C1 |
C3 |
H8 |
121.252 |
| C2 |
C1 |
C3 |
123.990 |
|
C2 |
C1 |
H5 |
116.208 |
| C2 |
C4 |
H9 |
121.782 |
|
C2 |
C4 |
H10 |
121.252 |
| C3 |
C1 |
H5 |
119.798 |
|
C4 |
C2 |
H6 |
119.798 |
| H7 |
C3 |
H8 |
116.959 |
|
H9 |
C4 |
H10 |
116.959 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.230 |
|
|
|
| 2 |
C |
-0.230 |
|
|
|
| 3 |
C |
-0.381 |
|
|
|
| 4 |
C |
-0.381 |
|
|
|
| 5 |
H |
0.210 |
|
|
|
| 6 |
H |
0.210 |
|
|
|
| 7 |
H |
0.201 |
|
|
|
| 8 |
H |
0.199 |
|
|
|
| 9 |
H |
0.201 |
|
|
|
| 10 |
H |
0.199 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.052 |
0.052 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-28.094 |
0.825 |
0.000 |
| y |
0.825 |
-22.819 |
0.000 |
| z |
0.000 |
0.000 |
-22.887 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.241 |
0.825 |
0.000 |
| y |
0.825 |
2.671 |
0.000 |
| z |
0.000 |
0.000 |
2.569 |
|
| Polar |
|---|
| 3z2-r2 | 5.139 |
|---|
| x2-y2 | -5.275 |
|---|
| xy | 0.825 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.240 |
0.053 |
0.000 |
| y |
0.053 |
7.949 |
0.000 |
| z |
0.000 |
0.000 |
6.142 |
<r2> (average value of r
2) Å
2
| <r2> |
84.918 |
| (<r2>)1/2 |
9.215 |