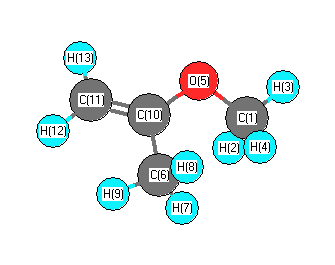
Vibrational Frequencies calculated at M06-2X/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3303 |
3129 |
2.95 |
|
|
|
| 2 |
A |
3215 |
3045 |
0.32 |
|
|
|
| 3 |
A |
3171 |
3003 |
21.52 |
|
|
|
| 4 |
A |
3152 |
2986 |
7.70 |
|
|
|
| 5 |
A |
3142 |
2977 |
24.89 |
|
|
|
| 6 |
A |
3113 |
2949 |
7.76 |
|
|
|
| 7 |
A |
3067 |
2905 |
34.28 |
|
|
|
| 8 |
A |
3043 |
2883 |
6.37 |
|
|
|
| 9 |
A |
1763 |
1670 |
94.54 |
|
|
|
| 10 |
A |
1587 |
1503 |
12.14 |
|
|
|
| 11 |
A |
1573 |
1490 |
12.84 |
|
|
|
| 12 |
A |
1553 |
1471 |
13.53 |
|
|
|
| 13 |
A |
1546 |
1464 |
11.02 |
|
|
|
| 14 |
A |
1521 |
1441 |
0.29 |
|
|
|
| 15 |
A |
1478 |
1400 |
3.11 |
|
|
|
| 16 |
A |
1463 |
1386 |
19.28 |
|
|
|
| 17 |
A |
1321 |
1252 |
158.51 |
|
|
|
| 18 |
A |
1207 |
1144 |
8.27 |
|
|
|
| 19 |
A |
1179 |
1117 |
3.68 |
|
|
|
| 20 |
A |
1125 |
1066 |
80.73 |
|
|
|
| 21 |
A |
1108 |
1050 |
8.23 |
|
|
|
| 22 |
A |
1060 |
1004 |
1.97 |
|
|
|
| 23 |
A |
959 |
909 |
77.86 |
|
|
|
| 24 |
A |
941 |
891 |
3.69 |
|
|
|
| 25 |
A |
803 |
761 |
8.15 |
|
|
|
| 26 |
A |
781 |
739 |
2.50 |
|
|
|
| 27 |
A |
543 |
515 |
6.50 |
|
|
|
| 28 |
A |
477 |
452 |
5.13 |
|
|
|
| 29 |
A |
427 |
404 |
0.70 |
|
|
|
| 30 |
A |
312 |
295 |
1.78 |
|
|
|
| 31 |
A |
248 |
235 |
1.63 |
|
|
|
| 32 |
A |
223 |
211 |
1.31 |
|
|
|
| 33 |
A |
81 |
77 |
9.97 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 25243.5 cm
-1
Scaled (by 0.9472) Zero Point Vibrational Energy (zpe) 23910.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.326 |
|
|
|
| 2 |
H |
0.194 |
|
|
|
| 3 |
H |
0.225 |
|
|
|
| 4 |
H |
0.192 |
|
|
|
| 5 |
O |
-0.546 |
|
|
|
| 6 |
C |
-0.628 |
|
|
|
| 7 |
H |
0.211 |
|
|
|
| 8 |
H |
0.220 |
|
|
|
| 9 |
H |
0.217 |
|
|
|
| 10 |
C |
0.272 |
|
|
|
| 11 |
C |
-0.428 |
|
|
|
| 12 |
H |
0.188 |
|
|
|
| 13 |
H |
0.209 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.843 |
1.765 |
0.777 |
2.105 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.374 |
0.295 |
0.515 |
| y |
0.295 |
-31.566 |
-0.680 |
| z |
0.515 |
-0.680 |
-33.548 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.183 |
0.295 |
0.515 |
| y |
0.295 |
-1.105 |
-0.680 |
| z |
0.515 |
-0.680 |
-4.078 |
|
| Polar |
|---|
| 3z2-r2 | -8.156 |
|---|
| x2-y2 | 4.192 |
|---|
| xy | 0.295 |
|---|
| xz | 0.515 |
|---|
| yz | -0.680 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.446 |
0.994 |
-0.322 |
| y |
0.994 |
6.496 |
-0.141 |
| z |
-0.322 |
-0.141 |
3.956 |
<r2> (average value of r
2) Å
2
| <r2> |
130.145 |
| (<r2>)1/2 |
11.408 |