Vibrational Frequencies calculated at M06-2X/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3595 |
3405 |
1.79 |
|
|
|
| 2 |
A |
3139 |
2973 |
4.59 |
|
|
|
| 3 |
A |
3124 |
2959 |
36.02 |
|
|
|
| 4 |
A |
3119 |
2954 |
29.69 |
|
|
|
| 5 |
A |
3103 |
2939 |
20.90 |
|
|
|
| 6 |
A |
3095 |
2931 |
1.06 |
|
|
|
| 7 |
A |
3066 |
2904 |
51.22 |
|
|
|
| 8 |
A |
3052 |
2891 |
2.01 |
|
|
|
| 9 |
A |
3048 |
2887 |
18.53 |
|
|
|
| 10 |
A |
3038 |
2878 |
9.02 |
|
|
|
| 11 |
A |
3035 |
2875 |
21.71 |
|
|
|
| 12 |
A |
3005 |
2846 |
32.36 |
|
|
|
| 13 |
A |
1586 |
1502 |
2.21 |
|
|
|
| 14 |
A |
1575 |
1492 |
8.82 |
|
|
|
| 15 |
A |
1573 |
1490 |
11.75 |
|
|
|
| 16 |
A |
1565 |
1482 |
8.14 |
|
|
|
| 17 |
A |
1556 |
1474 |
10.13 |
|
|
|
| 18 |
A |
1548 |
1467 |
3.57 |
|
|
|
| 19 |
A |
1479 |
1401 |
3.09 |
|
|
|
| 20 |
A |
1466 |
1389 |
10.92 |
|
|
|
| 21 |
A |
1445 |
1369 |
12.78 |
|
|
|
| 22 |
A |
1423 |
1348 |
1.04 |
|
|
|
| 23 |
A |
1394 |
1320 |
0.64 |
|
|
|
| 24 |
A |
1375 |
1303 |
0.58 |
|
|
|
| 25 |
A |
1341 |
1270 |
7.72 |
|
|
|
| 26 |
A |
1293 |
1224 |
0.08 |
|
|
|
| 27 |
A |
1271 |
1204 |
31.00 |
|
|
|
| 28 |
A |
1218 |
1154 |
0.64 |
|
|
|
| 29 |
A |
1191 |
1128 |
7.00 |
|
|
|
| 30 |
A |
1148 |
1087 |
4.42 |
|
|
|
| 31 |
A |
1075 |
1018 |
31.63 |
|
|
|
| 32 |
A |
1054 |
999 |
29.43 |
|
|
|
| 33 |
A |
1049 |
994 |
28.01 |
|
|
|
| 34 |
A |
1021 |
967 |
9.10 |
|
|
|
| 35 |
A |
964 |
913 |
2.97 |
|
|
|
| 36 |
A |
933 |
884 |
2.47 |
|
|
|
| 37 |
A |
850 |
805 |
4.13 |
|
|
|
| 38 |
A |
790 |
749 |
8.21 |
|
|
|
| 39 |
A |
507 |
481 |
5.82 |
|
|
|
| 40 |
A |
450 |
427 |
2.42 |
|
|
|
| 41 |
A |
406 |
384 |
4.59 |
|
|
|
| 42 |
A |
326 |
309 |
128.40 |
|
|
|
| 43 |
A |
312 |
296 |
17.13 |
|
|
|
| 44 |
A |
264 |
250 |
1.33 |
|
|
|
| 45 |
A |
248 |
235 |
2.54 |
|
|
|
| 46 |
A |
233 |
221 |
0.95 |
|
|
|
| 47 |
A |
125 |
118 |
5.28 |
|
|
|
| 48 |
A |
60 |
57 |
1.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36764.8 cm
-1
Scaled (by 0.9472) Zero Point Vibrational Energy (zpe) 34823.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
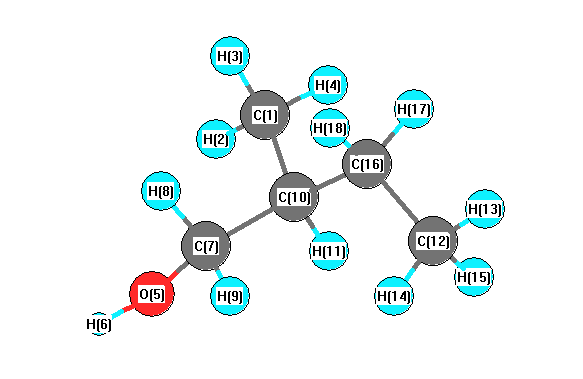
Charges from optimized geometry at M06-2X/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.541 |
|
|
|
| 2 |
H |
0.226 |
|
|
|
| 3 |
H |
0.177 |
|
|
|
| 4 |
H |
0.185 |
|
|
|
| 5 |
O |
-0.581 |
|
|
|
| 6 |
H |
0.351 |
|
|
|
| 7 |
C |
-0.106 |
|
|
|
| 8 |
H |
0.169 |
|
|
|
| 9 |
H |
0.182 |
|
|
|
| 10 |
C |
-0.290 |
|
|
|
| 11 |
H |
0.210 |
|
|
|
| 12 |
C |
-0.576 |
|
|
|
| 13 |
H |
0.199 |
|
|
|
| 14 |
H |
0.190 |
|
|
|
| 15 |
H |
0.196 |
|
|
|
| 16 |
C |
-0.382 |
|
|
|
| 17 |
H |
0.202 |
|
|
|
| 18 |
H |
0.188 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.447 |
-1.252 |
1.088 |
1.718 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.800 |
3.414 |
-2.705 |
| y |
3.414 |
-37.122 |
-1.627 |
| z |
-2.705 |
-1.627 |
-39.988 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.245 |
3.414 |
-2.705 |
| y |
3.414 |
2.772 |
-1.627 |
| z |
-2.705 |
-1.627 |
-1.527 |
|
| Polar |
|---|
| 3z2-r2 | -3.053 |
|---|
| x2-y2 | -2.678 |
|---|
| xy | 3.414 |
|---|
| xz | -2.705 |
|---|
| yz | -1.627 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.818 |
0.117 |
-0.050 |
| y |
0.117 |
7.954 |
-0.159 |
| z |
-0.050 |
-0.159 |
6.987 |
<r2> (average value of r
2) Å
2
| <r2> |
214.364 |
| (<r2>)1/2 |
14.641 |