Jump to
S1C2
Vibrational Frequencies calculated at B2PLYP=FULL/STO-3G
Geometric Data calculated at B2PLYP=FULL/STO-3G
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B2PLYP=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -340.526914 |
| Energy at 298.15K | -340.543612 |
| HF Energy | -340.382931 |
| Nuclear repulsion energy | 414.275457 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3488 |
3488 |
0.00 |
|
|
|
| 2 |
A1 |
3377 |
3377 |
0.00 |
|
|
|
| 3 |
A1 |
1683 |
1683 |
0.00 |
|
|
|
| 4 |
A1 |
1472 |
1472 |
0.00 |
|
|
|
| 5 |
A1 |
1382 |
1382 |
0.00 |
|
|
|
| 6 |
A1 |
1084 |
1084 |
0.00 |
|
|
|
| 7 |
A1 |
1072 |
1072 |
0.00 |
|
|
|
| 8 |
A1 |
830 |
830 |
0.00 |
|
|
|
| 9 |
A1 |
610 |
610 |
0.00 |
|
|
|
| 10 |
A1 |
93 |
93 |
0.00 |
|
|
|
| 11 |
A2 |
3495 |
3495 |
0.00 |
|
|
|
| 12 |
A2 |
3374 |
3374 |
10.86 |
|
|
|
| 13 |
A2 |
1672 |
1672 |
0.00 |
|
|
|
| 14 |
A2 |
1514 |
1514 |
9.48 |
|
|
|
| 15 |
A2 |
1279 |
1279 |
0.00 |
|
|
|
| 16 |
A2 |
1023 |
1023 |
8.09 |
|
|
|
| 17 |
A2 |
866 |
866 |
0.00 |
|
|
|
| 18 |
A2 |
777 |
777 |
33.95 |
|
|
|
| 19 |
E |
3498 |
3498 |
3.85 |
|
|
|
| 19 |
E |
3498 |
3498 |
3.85 |
|
|
|
| 20 |
E |
3489 |
3489 |
0.00 |
|
|
|
| 20 |
E |
3489 |
3489 |
0.00 |
|
|
|
| 21 |
E |
3376 |
3376 |
8.92 |
|
|
|
| 21 |
E |
3376 |
3376 |
8.92 |
|
|
|
| 22 |
E |
3374 |
3374 |
0.00 |
|
|
|
| 22 |
E |
3374 |
3374 |
0.00 |
|
|
|
| 23 |
E |
1676 |
1676 |
0.81 |
|
|
|
| 23 |
E |
1676 |
1676 |
0.81 |
|
|
|
| 24 |
E |
1667 |
1667 |
0.00 |
|
|
|
| 24 |
E |
1667 |
1667 |
0.00 |
|
|
|
| 25 |
E |
1481 |
1481 |
0.00 |
|
|
|
| 25 |
E |
1481 |
1481 |
0.00 |
|
|
|
| 26 |
E |
1456 |
1456 |
0.03 |
|
|
|
| 26 |
E |
1456 |
1456 |
0.03 |
|
|
|
| 27 |
E |
1419 |
1419 |
0.10 |
|
|
|
| 27 |
E |
1419 |
1419 |
0.10 |
|
|
|
| 28 |
E |
1417 |
1417 |
0.00 |
|
|
|
| 28 |
E |
1417 |
1417 |
0.00 |
|
|
|
| 29 |
E |
1295 |
1295 |
0.00 |
|
|
|
| 29 |
E |
1295 |
1295 |
0.00 |
|
|
|
| 30 |
E |
1178 |
1178 |
5.20 |
|
|
|
| 30 |
E |
1178 |
1178 |
5.20 |
|
|
|
| 31 |
E |
1106 |
1106 |
0.00 |
|
|
|
| 31 |
E |
1106 |
1106 |
0.00 |
|
|
|
| 32 |
E |
993 |
993 |
2.35 |
|
|
|
| 32 |
E |
993 |
993 |
2.35 |
|
|
|
| 33 |
E |
898 |
898 |
4.61 |
|
|
|
| 33 |
E |
898 |
898 |
4.61 |
|
|
|
| 34 |
E |
598 |
598 |
0.00 |
|
|
|
| 34 |
E |
598 |
598 |
0.00 |
|
|
|
| 35 |
E |
427 |
427 |
0.29 |
|
|
|
| 35 |
E |
427 |
427 |
0.29 |
|
|
|
| 36 |
E |
323 |
323 |
0.00 |
|
|
|
| 36 |
E |
323 |
323 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 44214.3 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 44214.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/STO-3G
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.365 |
| N2 |
0.000 |
0.000 |
-1.365 |
| C3 |
-0.002 |
1.413 |
0.791 |
| C4 |
1.224 |
-0.705 |
0.791 |
| C5 |
-1.222 |
-0.708 |
0.791 |
| C6 |
0.002 |
1.413 |
-0.791 |
| C7 |
-1.224 |
-0.705 |
-0.791 |
| C8 |
1.222 |
-0.708 |
-0.791 |
| H9 |
0.891 |
1.930 |
1.183 |
| H10 |
-0.899 |
1.926 |
1.179 |
| H11 |
1.226 |
-1.737 |
1.183 |
| H12 |
2.117 |
-0.184 |
1.179 |
| H13 |
-2.117 |
-0.193 |
1.183 |
| H14 |
-1.218 |
-1.742 |
1.179 |
| H15 |
-0.891 |
1.930 |
-1.183 |
| H16 |
0.899 |
1.926 |
-1.179 |
| H17 |
-1.226 |
-1.737 |
-1.183 |
| H18 |
-2.117 |
-0.184 |
-1.179 |
| H19 |
2.117 |
-0.193 |
-1.183 |
| H20 |
1.218 |
-1.742 |
-1.179 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.7300 | 1.5248 | 1.5248 | 1.5248 | 2.5776 | 2.5776 | 2.5776 | 2.1336 | 2.1334 | 2.1336 | 2.1334 | 2.1336 | 2.1334 | 3.3183 | 3.3148 | 3.3183 | 3.3148 | 3.3183 | 3.3148 |
N2 | 2.7300 | | 2.5776 | 2.5776 | 2.5776 | 1.5248 | 1.5248 | 1.5248 | 3.3183 | 3.3148 | 3.3183 | 3.3148 | 3.3183 | 3.3148 | 2.1336 | 2.1334 | 2.1336 | 2.1334 | 2.1336 | 2.1334 | C3 | 1.5248 | 2.5776 | | 2.4468 | 2.4468 | 1.5821 | 2.9122 | 2.9153 | 1.1041 | 1.1041 | 3.4030 | 2.6814 | 2.6844 | 3.4028 | 2.2261 | 2.2262 | 3.9133 | 3.3022 | 3.3113 | 3.9139 | C4 | 1.5248 | 2.5776 | 2.4468 | | 2.4468 | 2.9122 | 2.9153 | 1.5821 | 2.6844 | 3.4028 | 1.1041 | 1.1041 | 3.4030 | 2.6814 | 3.9133 | 3.3022 | 3.3113 | 3.9139 | 2.2261 | 2.2262 | C5 | 1.5248 | 2.5776 | 2.4468 | 2.4468 | | 2.9153 | 1.5821 | 2.9122 | 3.4030 | 2.6814 | 2.6844 | 3.4028 | 1.1041 | 1.1041 | 3.3113 | 3.9139 | 2.2261 | 2.2262 | 3.9133 | 3.3022 | C6 | 2.5776 | 1.5248 | 1.5821 | 2.9122 | 2.9153 | | 2.4468 | 2.4468 | 2.2261 | 2.2262 | 3.9133 | 3.3022 | 3.3113 | 3.9139 | 1.1041 | 1.1041 | 3.4030 | 2.6814 | 2.6844 | 3.4028 | C7 | 2.5776 | 1.5248 | 2.9122 | 2.9153 | 1.5821 | 2.4468 | | 2.4468 | 3.9133 | 3.3022 | 3.3113 | 3.9139 | 2.2261 | 2.2262 | 2.6844 | 3.4028 | 1.1041 | 1.1041 | 3.4030 | 2.6814 | C8 | 2.5776 | 1.5248 | 2.9153 | 1.5821 | 2.9122 | 2.4468 | 2.4468 | | 3.3113 | 3.9139 | 2.2261 | 2.2262 | 3.9133 | 3.3022 | 3.4030 | 2.6814 | 2.6844 | 3.4028 | 1.1041 | 1.1041 | H9 | 2.1336 | 3.3183 | 1.1041 | 2.6844 | 3.4030 | 2.2261 | 3.9133 | 3.3113 | | 1.7908 | 3.6821 | 2.4436 | 3.6821 | 4.2343 | 2.9624 | 2.3619 | 4.8503 | 4.3702 | 3.4068 | 4.3778 | H10 | 2.1334 | 3.3148 | 1.1041 | 3.4028 | 2.6814 | 2.2262 | 3.3022 | 3.9139 | 1.7908 | | 4.2343 | 3.6810 | 2.4436 | 3.6810 | 2.3619 | 2.9656 | 4.3702 | 3.3900 | 4.3778 | 4.8467 | H11 | 2.1336 | 3.3183 | 3.4030 | 1.1041 | 2.6844 | 3.9133 | 3.3113 | 2.2261 | 3.6821 | 4.2343 | | 1.7908 | 3.6821 | 2.4436 | 4.8503 | 4.3702 | 3.4068 | 4.3778 | 2.9624 | 2.3619 | H12 | 2.1334 | 3.3148 | 2.6814 | 1.1041 | 3.4028 | 3.3022 | 3.9139 | 2.2262 | 2.4436 | 3.6810 | 1.7908 | | 4.2343 | 3.6810 | 4.3702 | 3.3900 | 4.3778 | 4.8467 | 2.3619 | 2.9656 | H13 | 2.1336 | 3.3183 | 2.6844 | 3.4030 | 1.1041 | 3.3113 | 2.2261 | 3.9133 | 3.6821 | 2.4436 | 3.6821 | 4.2343 | | 1.7908 | 3.4068 | 4.3778 | 2.9624 | 2.3619 | 4.8503 | 4.3702 | H14 | 2.1334 | 3.3148 | 3.4028 | 2.6814 | 1.1041 | 3.9139 | 2.2262 | 3.3022 | 4.2343 | 3.6810 | 2.4436 | 3.6810 | 1.7908 | | 4.3778 | 4.8467 | 2.3619 | 2.9656 | 4.3702 | 3.3900 | H15 | 3.3183 | 2.1336 | 2.2261 | 3.9133 | 3.3113 | 1.1041 | 2.6844 | 3.4030 | 2.9624 | 2.3619 | 4.8503 | 4.3702 | 3.4068 | 4.3778 | | 1.7908 | 3.6821 | 2.4436 | 3.6821 | 4.2343 | H16 | 3.3148 | 2.1334 | 2.2262 | 3.3022 | 3.9139 | 1.1041 | 3.4028 | 2.6814 | 2.3619 | 2.9656 | 4.3702 | 3.3900 | 4.3778 | 4.8467 | 1.7908 | | 4.2343 | 3.6810 | 2.4436 | 3.6810 | H17 | 3.3183 | 2.1336 | 3.9133 | 3.3113 | 2.2261 | 3.4030 | 1.1041 | 2.6844 | 4.8503 | 4.3702 | 3.4068 | 4.3778 | 2.9624 | 2.3619 | 3.6821 | 4.2343 | | 1.7908 | 3.6821 | 2.4436 | H18 | 3.3148 | 2.1334 | 3.3022 | 3.9139 | 2.2262 | 2.6814 | 1.1041 | 3.4028 | 4.3702 | 3.3900 | 4.3778 | 4.8467 | 2.3619 | 2.9656 | 2.4436 | 3.6810 | 1.7908 | | 4.2343 | 3.6810 | H19 | 3.3183 | 2.1336 | 3.3113 | 2.2261 | 3.9133 | 2.6844 | 3.4030 | 1.1041 | 3.4068 | 4.3778 | 2.9624 | 2.3619 | 4.8503 | 4.3702 | 3.6821 | 2.4436 | 3.6821 | 4.2343 | | 1.7908 | H20 | 3.3148 | 2.1334 | 3.9139 | 2.2262 | 3.3022 | 3.4028 | 2.6814 | 1.1041 | 4.3778 | 4.8467 | 2.3619 | 2.9656 | 4.3702 | 3.3900 | 4.2343 | 3.6810 | 2.4436 | 3.6810 | 1.7908 | |
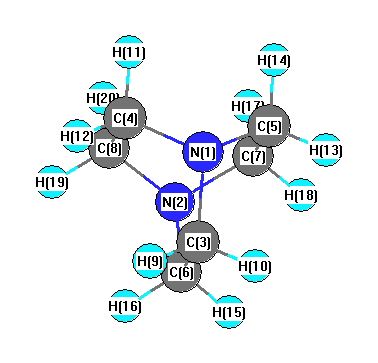 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
112.111 |
|
N1 |
C3 |
H9 |
107.424 |
| N1 |
C3 |
H10 |
107.406 |
|
N1 |
C4 |
C8 |
112.111 |
| N1 |
C4 |
H11 |
107.424 |
|
N1 |
C4 |
H12 |
107.406 |
| N1 |
C5 |
C7 |
112.111 |
|
N1 |
C5 |
H13 |
107.424 |
| N1 |
C5 |
H14 |
107.406 |
|
N2 |
C6 |
C3 |
112.111 |
| N2 |
C6 |
H15 |
107.424 |
|
N2 |
C6 |
H16 |
107.406 |
| N2 |
C7 |
C5 |
112.111 |
|
N2 |
C7 |
H17 |
107.424 |
| N2 |
C7 |
H18 |
107.406 |
|
N2 |
C8 |
C4 |
112.111 |
| N2 |
C8 |
H19 |
107.424 |
|
N2 |
C8 |
H20 |
107.406 |
| C3 |
N1 |
C4 |
106.707 |
|
C3 |
N1 |
C5 |
106.707 |
| C3 |
C6 |
H15 |
110.674 |
|
C3 |
C6 |
H16 |
110.683 |
| C4 |
N1 |
C5 |
106.707 |
|
C4 |
C8 |
H19 |
110.674 |
| C4 |
C8 |
H20 |
110.683 |
|
C5 |
C6 |
H15 |
101.166 |
| C5 |
C6 |
H16 |
150.451 |
|
C6 |
N2 |
C7 |
106.707 |
| C6 |
N2 |
C8 |
106.707 |
|
C6 |
C3 |
H9 |
110.674 |
| C6 |
C3 |
H10 |
110.683 |
|
C7 |
N2 |
C8 |
106.707 |
| C7 |
C5 |
H13 |
110.674 |
|
C7 |
C5 |
H14 |
110.683 |
| C8 |
C4 |
H11 |
110.674 |
|
C8 |
C4 |
H12 |
110.683 |
| H9 |
C3 |
H10 |
108.381 |
|
H11 |
C4 |
H12 |
108.381 |
| H13 |
C5 |
H14 |
108.381 |
|
H15 |
C6 |
H16 |
108.381 |
| H17 |
C7 |
H18 |
108.381 |
|
H19 |
C8 |
H20 |
108.381 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability