Jump to
S1C2
Energy calculated at B2PLYP=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -275.307261 |
| Energy at 298.15K | |
| HF Energy | -275.072924 |
| Nuclear repulsion energy | 117.279740 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2112 |
2112 |
37.93 |
42.76 |
0.15 |
0.27 |
| 2 |
A1 |
747 |
747 |
10.96 |
9.84 |
0.11 |
0.21 |
| 3 |
A1 |
519 |
519 |
83.90 |
0.53 |
0.26 |
0.41 |
| 4 |
A1 |
82 |
82 |
9.04 |
1.85 |
0.64 |
0.78 |
| 5 |
A2 |
459 |
459 |
0.00 |
2.94 |
0.75 |
0.86 |
| 6 |
B1 |
481 |
481 |
104.48 |
0.19 |
0.75 |
0.86 |
| 7 |
B2 |
2132 |
2132 |
1077.70 |
2.78 |
0.75 |
0.86 |
| 8 |
B2 |
1251 |
1251 |
88.96 |
0.85 |
0.75 |
0.86 |
| 9 |
B2 |
451 |
451 |
11.00 |
1.99 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4117.6 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 4117.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/6-31G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.521 |
| B2 |
0.000 |
1.247 |
0.067 |
| B3 |
0.000 |
-1.247 |
0.067 |
| O4 |
0.000 |
2.404 |
-0.302 |
| O5 |
0.000 |
-2.404 |
-0.302 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3274 | 1.3274 | 2.5414 | 2.5414 |
B2 | 1.3274 | | 2.4950 | 1.2145 | 3.6706 | B3 | 1.3274 | 2.4950 | | 3.6706 | 1.2145 | O4 | 2.5414 | 1.2145 | 3.6706 | | 4.8090 | O5 | 2.5414 | 3.6706 | 1.2145 | 4.8090 | |
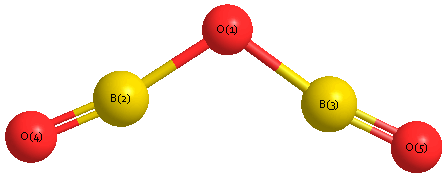 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.706 |
|
O1 |
B3 |
O5 |
177.706 |
| B2 |
O1 |
B3 |
140.021 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B2PLYP=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -275.306117 |
| Energy at 298.15K | |
| HF Energy | -275.072222 |
| Nuclear repulsion energy | 116.632416 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2120 |
2120 |
0.00 |
62.77 |
0.18 |
0.31 |
| 2 |
Σg |
679 |
679 |
0.00 |
10.63 |
0.17 |
0.29 |
| 3 |
Σu |
2170 |
2170 |
1436.59 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1329 |
1329 |
84.57 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
465 |
465 |
0.00 |
3.46 |
0.75 |
0.86 |
| 5 |
Πg |
465 |
465 |
0.00 |
3.46 |
0.75 |
0.86 |
| 6 |
Πu |
457 |
457 |
112.62 |
0.00 |
0.00 |
0.00 |
| 6 |
Πu |
457 |
457 |
112.62 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
68i |
68i |
4.34 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
68i |
68i |
4.34 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 4003.8 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 4003.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/6-31G**
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.313 |
| B3 |
0.000 |
0.000 |
-1.313 |
| O4 |
0.000 |
0.000 |
2.528 |
| O5 |
0.000 |
0.000 |
-2.528 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3126 | 1.3126 | 2.5281 | 2.5281 |
B2 | 1.3126 | | 2.6251 | 1.2156 | 3.8407 | B3 | 1.3126 | 2.6251 | | 3.8407 | 1.2156 | O4 | 2.5281 | 1.2156 | 3.8407 | | 5.0563 | O5 | 2.5281 | 3.8407 | 1.2156 | 5.0563 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability