Jump to
S1C2
S1C3
Energy calculated at B2PLYP=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -1592.083883 |
| Energy at 298.15K | |
| HF Energy | -1591.908001 |
| Nuclear repulsion energy | 346.052525 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
493 |
493 |
0.00 |
51.66 |
0.07 |
0.13 |
| 2 |
A1 |
197 |
197 |
0.00 |
3.00 |
0.75 |
0.86 |
| 3 |
B1 |
432 |
432 |
0.00 |
34.47 |
0.75 |
0.86 |
| 4 |
B2 |
314 |
314 |
0.30 |
11.20 |
0.75 |
0.86 |
| 5 |
E |
409 |
409 |
0.07 |
7.29 |
0.75 |
0.86 |
| 5 |
E |
409 |
409 |
0.07 |
7.29 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1126.9 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 1126.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/6-31G**
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.451 |
0.313 |
| S2 |
0.000 |
-1.451 |
0.313 |
| S3 |
-1.451 |
0.000 |
-0.313 |
| S4 |
1.451 |
0.000 |
-0.313 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.9011 | 2.1447 | 2.1447 |
S2 | 2.9011 | | 2.1447 | 2.1447 | S3 | 2.1447 | 2.1447 | | 2.9011 | S4 | 2.1447 | 2.1447 | 2.9011 | |
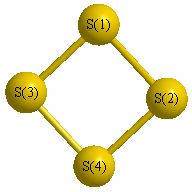 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
47.442 |
|
S1 |
S3 |
S4 |
47.442 |
| S2 |
S1 |
S3 |
47.442 |
|
S2 |
S4 |
S3 |
47.442 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at B2PLYP=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -1592.130761 |
| Energy at 298.15K | |
| HF Energy | -1591.902074 |
| Nuclear repulsion energy | 323.048014 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
679 |
679 |
0.00 |
38.80 |
0.22 |
0.36 |
| 2 |
A1 |
215 |
215 |
0.00 |
33.49 |
0.44 |
0.61 |
| 3 |
A1 |
108 |
108 |
6.86 |
0.00 |
0.20 |
0.34 |
| 4 |
A2 |
214 |
214 |
0.00 |
0.00 |
0.75 |
0.86 |
| 5 |
B2 |
667 |
667 |
39.26 |
0.00 |
0.75 |
0.86 |
| 6 |
B2 |
295 |
295 |
0.00 |
18.59 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1088.2 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 1088.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/6-31G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.346 |
0.964 |
| S2 |
0.000 |
-1.346 |
0.964 |
| S3 |
0.000 |
1.345 |
-0.964 |
| S4 |
0.000 |
-1.345 |
-0.964 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.6912 | 1.9282 | 3.3103 |
S2 | 2.6912 | | 3.3103 | 1.9282 | S3 | 1.9282 | 3.3103 | | 2.6903 | S4 | 3.3103 | 1.9282 | 2.6903 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
89.986 |
|
S1 |
S3 |
S4 |
90.014 |
| S2 |
S1 |
S3 |
89.986 |
|
S2 |
S4 |
S3 |
90.014 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at B2PLYP=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -1592.130761 |
| Energy at 298.15K | |
| HF Energy | -1591.902075 |
| Nuclear repulsion energy | 323.050964 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
678 |
678 |
0.00 |
38.80 |
0.22 |
0.36 |
| 2 |
Ag |
215 |
215 |
0.00 |
33.39 |
0.44 |
0.61 |
| 3 |
Au |
214 |
214 |
0.00 |
0.00 |
0.00 |
0.00 |
| 4 |
B1u |
667 |
667 |
39.27 |
0.00 |
0.00 |
0.00 |
| 5 |
B2u |
108 |
108 |
6.86 |
0.00 |
0.00 |
0.00 |
| 6 |
B3g |
295 |
295 |
0.00 |
18.59 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1088.2 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 1088.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/6-31G**
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.964 |
1.345 |
| S2 |
0.000 |
0.964 |
-1.345 |
| S3 |
0.000 |
-0.964 |
1.345 |
| S4 |
0.000 |
-0.964 |
-1.345 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.6907 | 1.9283 | 3.3103 |
S2 | 2.6907 | | 3.3103 | 1.9283 | S3 | 1.9283 | 3.3103 | | 2.6907 | S4 | 3.3103 | 1.9283 | 2.6907 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
90.000 |
|
S1 |
S3 |
S4 |
90.000 |
| S2 |
S1 |
S3 |
90.000 |
|
S2 |
S4 |
S3 |
90.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability