Jump to
S1C2
Energy calculated at B2PLYP=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1194.271485 |
| Energy at 298.15K | -1194.271884 |
| HF Energy | -1194.054947 |
| Nuclear repulsion energy | 180.605506 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at B2PLYP=FULL/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.000 |
0.668 |
| S2 |
0.000 |
1.659 |
-0.334 |
| S3 |
0.000 |
-1.659 |
-0.334 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
| S1 | | 1.9384 | 1.9384 |
S2 | 1.9384 | | 3.3178 | S3 | 1.9384 | 3.3178 | |
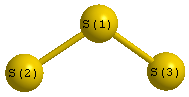 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S2 |
S1 |
S3 |
117.700 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B2PLYP=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1194.259954 |
| Energy at 298.15K | -1194.260447 |
| HF Energy | -1194.054908 |
| Nuclear repulsion energy | 194.048727 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at B2PLYP=FULL/cc-pVTZ
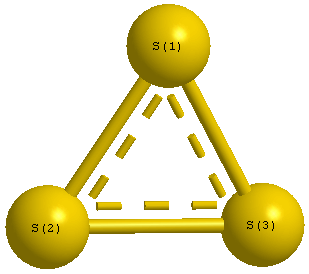
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.209 |
0.000 |
| S2 |
1.047 |
-0.605 |
0.000 |
| S3 |
-1.047 |
-0.605 |
0.000 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
| S1 | | 2.0944 | 2.0944 |
S2 | 2.0944 | | 2.0944 | S3 | 2.0944 | 2.0944 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S2 |
S1 |
S3 |
60.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability