Jump to
S1C2
Energy calculated at B2PLYP=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -275.485830 |
| Energy at 298.15K | |
| HF Energy | -275.181566 |
| Nuclear repulsion energy | 117.437318 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2090 |
1998 |
35.53 |
55.34 |
0.12 |
0.22 |
| 2 |
A1 |
739 |
707 |
9.80 |
10.11 |
0.08 |
0.15 |
| 3 |
A1 |
524 |
501 |
85.14 |
0.55 |
0.13 |
0.23 |
| 4 |
A1 |
88 |
84 |
11.31 |
1.46 |
0.61 |
0.75 |
| 5 |
A2 |
469 |
448 |
0.00 |
2.21 |
0.75 |
0.86 |
| 6 |
B1 |
486 |
464 |
105.35 |
0.13 |
0.75 |
0.86 |
| 7 |
B2 |
2101 |
2008 |
1167.89 |
2.03 |
0.75 |
0.86 |
| 8 |
B2 |
1252 |
1196 |
91.95 |
0.47 |
0.75 |
0.86 |
| 9 |
B2 |
460 |
440 |
10.50 |
1.69 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4104.4 cm
-1
Scaled (by 0.9558) Zero Point Vibrational Energy (zpe) 3923.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
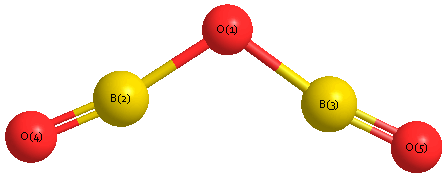
Geometric Data calculated at B2PLYP=FULL/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.489 |
| B2 |
0.000 |
1.253 |
0.061 |
| B3 |
0.000 |
-1.253 |
0.061 |
| O4 |
0.000 |
2.415 |
-0.282 |
| O5 |
0.000 |
-2.415 |
-0.282 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3245 | 1.3245 | 2.5346 | 2.5346 |
B2 | 1.3245 | | 2.5070 | 1.2106 | 3.6840 | B3 | 1.3245 | 2.5070 | | 3.6840 | 1.2106 | O4 | 2.5346 | 1.2106 | 3.6840 | | 4.8291 | O5 | 2.5346 | 3.6840 | 1.2106 | 4.8291 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.618 |
|
O1 |
B3 |
O5 |
177.618 |
| B2 |
O1 |
B3 |
142.317 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B2PLYP=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -275.484504 |
| Energy at 298.15K | |
| HF Energy | -275.180390 |
| Nuclear repulsion energy | 116.907160 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULL/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2094 |
2002 |
0.00 |
75.67 |
0.14 |
0.25 |
| 2 |
Σg |
676 |
646 |
0.00 |
10.90 |
0.13 |
0.22 |
| 3 |
Σu |
2134 |
2040 |
1509.87 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1322 |
1263 |
86.29 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
471 |
450 |
0.00 |
2.48 |
0.75 |
0.86 |
| 5 |
Πg |
471 |
450 |
0.00 |
2.48 |
0.75 |
0.86 |
| 6 |
Πu |
455 |
434 |
113.75 |
0.00 |
0.00 |
0.00 |
| 6 |
Πu |
455 |
434 |
113.75 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
85i |
81i |
4.75 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
85i |
81i |
4.75 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3953.8 cm
-1
Scaled (by 0.9558) Zero Point Vibrational Energy (zpe) 3779.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULL/cc-pVTZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.310 |
| B3 |
0.000 |
0.000 |
-1.310 |
| O4 |
0.000 |
0.000 |
2.522 |
| O5 |
0.000 |
0.000 |
-2.522 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3103 | 1.3103 | 2.5221 | 2.5221 |
B2 | 1.3103 | | 2.6207 | 1.2118 | 3.8325 | B3 | 1.3103 | 2.6207 | | 3.8325 | 1.2118 | O4 | 2.5221 | 1.2118 | 3.8325 | | 5.0443 | O5 | 2.5221 | 3.8325 | 1.2118 | 5.0443 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability