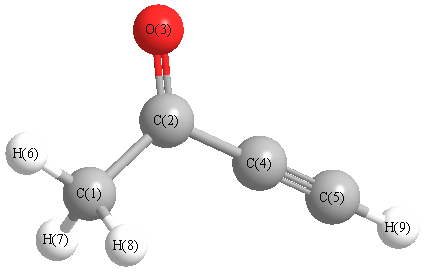
Vibrational Frequencies calculated at CCD/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3508 |
3326 |
45.85 |
|
|
|
| 2 |
A' |
3198 |
3031 |
8.46 |
|
|
|
| 3 |
A' |
3083 |
2922 |
2.11 |
|
|
|
| 4 |
A' |
2229 |
2113 |
43.49 |
|
|
|
| 5 |
A' |
1828 |
1733 |
154.78 |
|
|
|
| 6 |
A' |
1501 |
1423 |
12.08 |
|
|
|
| 7 |
A' |
1434 |
1359 |
28.99 |
|
|
|
| 8 |
A' |
1238 |
1174 |
134.97 |
|
|
|
| 9 |
A' |
1014 |
961 |
23.91 |
|
|
|
| 10 |
A' |
757 |
718 |
17.30 |
|
|
|
| 11 |
A' |
680 |
645 |
42.78 |
|
|
|
| 12 |
A' |
605 |
574 |
11.95 |
|
|
|
| 13 |
A' |
450 |
426 |
1.37 |
|
|
|
| 14 |
A' |
170 |
161 |
3.64 |
|
|
|
| 15 |
A" |
3157 |
2993 |
7.01 |
|
|
|
| 16 |
A" |
1495 |
1417 |
9.37 |
|
|
|
| 17 |
A" |
1063 |
1008 |
2.84 |
|
|
|
| 18 |
A" |
660 |
626 |
44.66 |
|
|
|
| 19 |
A" |
568 |
538 |
0.12 |
|
|
|
| 20 |
A" |
180 |
171 |
0.57 |
|
|
|
| 21 |
A" |
128 |
121 |
0.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14471.6 cm
-1
Scaled (by 0.948) Zero Point Vibrational Energy (zpe) 13719.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.