Jump to
S1C2
Vibrational Frequencies calculated at CCD/TZVP
Geometric Data calculated at CCD/TZVP
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCD/TZVP
| | hartrees |
|---|
| Energy at 0K | -322.904635 |
| Energy at 298.15K | -322.913726 |
| HF Energy | -321.849630 |
| Nuclear repulsion energy | 242.423152 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCD/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3160 |
2995 |
32.59 |
|
|
|
| 2 |
A |
3153 |
2989 |
38.57 |
|
|
|
| 3 |
A |
3147 |
2984 |
26.18 |
|
|
|
| 4 |
A |
3122 |
2960 |
12.58 |
|
|
|
| 5 |
A |
3094 |
2933 |
28.88 |
|
|
|
| 6 |
A |
3078 |
2918 |
16.64 |
|
|
|
| 7 |
A |
3072 |
2913 |
21.33 |
|
|
|
| 8 |
A |
1782 |
1689 |
177.49 |
|
|
|
| 9 |
A |
1539 |
1459 |
8.11 |
|
|
|
| 10 |
A |
1534 |
1455 |
3.67 |
|
|
|
| 11 |
A |
1522 |
1443 |
5.86 |
|
|
|
| 12 |
A |
1510 |
1432 |
1.39 |
|
|
|
| 13 |
A |
1463 |
1387 |
12.13 |
|
|
|
| 14 |
A |
1449 |
1374 |
9.28 |
|
|
|
| 15 |
A |
1419 |
1346 |
1.04 |
|
|
|
| 16 |
A |
1339 |
1269 |
13.33 |
|
|
|
| 17 |
A |
1323 |
1254 |
4.15 |
|
|
|
| 18 |
A |
1222 |
1158 |
13.03 |
|
|
|
| 19 |
A |
1153 |
1093 |
30.74 |
|
|
|
| 20 |
A |
1124 |
1066 |
27.78 |
|
|
|
| 21 |
A |
1037 |
983 |
9.02 |
|
|
|
| 22 |
A |
959 |
909 |
144.78 |
|
|
|
| 23 |
A |
896 |
850 |
1.77 |
|
|
|
| 24 |
A |
884 |
838 |
345.32 |
|
|
|
| 25 |
A |
806 |
764 |
13.82 |
|
|
|
| 26 |
A |
652 |
618 |
17.45 |
|
|
|
| 27 |
A |
467 |
443 |
4.91 |
|
|
|
| 28 |
A |
398 |
378 |
2.15 |
|
|
|
| 29 |
A |
285 |
270 |
0.97 |
|
|
|
| 30 |
A |
244 |
231 |
0.85 |
|
|
|
| 31 |
A |
172 |
163 |
0.36 |
|
|
|
| 32 |
A |
116 |
110 |
1.12 |
|
|
|
| 33 |
A |
35 |
33 |
0.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23578.7 cm
-1
Scaled (by 0.948) Zero Point Vibrational Energy (zpe) 22352.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCD/TZVP
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.787 |
1.157 |
0.066 |
| C2 |
1.774 |
-0.346 |
-0.201 |
| C3 |
0.505 |
-1.024 |
0.287 |
| O4 |
-0.634 |
-0.533 |
-0.435 |
| N5 |
-1.516 |
0.123 |
0.423 |
| O6 |
-2.436 |
0.546 |
-0.164 |
| H7 |
2.732 |
1.598 |
-0.257 |
| H8 |
1.663 |
1.365 |
1.131 |
| H9 |
0.982 |
1.657 |
-0.475 |
| H10 |
1.889 |
-0.541 |
-1.270 |
| H11 |
2.617 |
-0.826 |
0.306 |
| H12 |
0.544 |
-2.102 |
0.121 |
| H13 |
0.347 |
-0.838 |
1.352 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5266 | 2.5399 | 2.9945 | 3.4793 | 4.2736 | 1.0910 | 1.0928 | 1.0909 | 2.1625 | 2.1628 | 3.4879 | 2.7765 |
C2 | 1.5266 | | 1.5194 | 2.4266 | 3.3812 | 4.3040 | 2.1680 | 2.1712 | 2.1712 | 1.0927 | 1.0941 | 2.1671 | 2.1655 | C3 | 2.5399 | 1.5194 | | 1.4353 | 2.3277 | 3.3646 | 3.4829 | 2.7860 | 2.8279 | 2.1384 | 2.1213 | 1.0909 | 1.0924 | O4 | 2.9945 | 2.4266 | 1.4353 | | 1.3941 | 2.1178 | 3.9871 | 3.3659 | 2.7216 | 2.6573 | 3.3470 | 2.0396 | 2.0611 | N5 | 3.4793 | 3.3812 | 2.3277 | 1.3941 | | 1.1709 | 4.5470 | 3.4855 | 3.0656 | 3.8596 | 4.2418 | 3.0474 | 2.2928 | O6 | 4.2736 | 4.3040 | 3.3646 | 2.1178 | 1.1709 | | 5.2744 | 4.3765 | 3.6079 | 4.5945 | 5.2572 | 3.9973 | 3.4585 | H7 | 1.0910 | 2.1680 | 3.4829 | 3.9871 | 4.5470 | 5.2744 | | 1.7672 | 1.7635 | 2.5123 | 2.4912 | 4.3143 | 3.7695 | H8 | 1.0928 | 2.1712 | 2.7860 | 3.3659 | 3.4855 | 4.3765 | 1.7672 | | 1.7690 | 3.0738 | 2.5279 | 3.7800 | 2.5756 | H9 | 1.0909 | 2.1712 | 2.8279 | 2.7216 | 3.0656 | 3.6079 | 1.7635 | 1.7690 | | 2.5065 | 3.0733 | 3.8304 | 3.1569 | H10 | 2.1625 | 1.0927 | 2.1384 | 2.6573 | 3.8596 | 4.5945 | 2.5123 | 3.0738 | 2.5065 | | 1.7590 | 2.4852 | 3.0560 | H11 | 2.1628 | 1.0941 | 2.1213 | 3.3470 | 4.2418 | 5.2572 | 2.4912 | 2.5279 | 3.0733 | 1.7590 | | 2.4404 | 2.4994 | H12 | 3.4879 | 2.1671 | 1.0909 | 2.0396 | 3.0474 | 3.9973 | 4.3143 | 3.7800 | 3.8304 | 2.4852 | 2.4404 | | 1.7753 | H13 | 2.7765 | 2.1655 | 1.0924 | 2.0611 | 2.2928 | 3.4585 | 3.7695 | 2.5756 | 3.1569 | 3.0560 | 2.4994 | 1.7753 | |
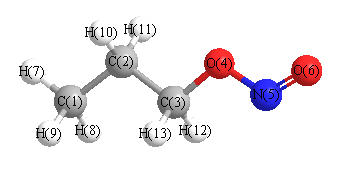 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.991 |
|
C1 |
C2 |
H10 |
110.193 |
| C1 |
C2 |
H11 |
110.142 |
|
C2 |
C1 |
H7 |
110.740 |
| C2 |
C1 |
H8 |
110.883 |
|
C2 |
C1 |
H9 |
110.995 |
| C2 |
C3 |
O4 |
110.389 |
|
C2 |
C3 |
H12 |
111.183 |
| C2 |
C3 |
H13 |
110.957 |
|
C3 |
C2 |
H10 |
108.800 |
| C3 |
C2 |
H11 |
107.405 |
|
C3 |
O4 |
N5 |
110.705 |
| O4 |
C3 |
H12 |
106.891 |
|
O4 |
C3 |
H13 |
108.491 |
| O4 |
N5 |
O6 |
111.016 |
|
H7 |
C1 |
H8 |
108.039 |
| H7 |
C1 |
H9 |
107.857 |
|
H8 |
C1 |
H9 |
108.207 |
| H10 |
C2 |
H11 |
107.101 |
|
H12 |
C3 |
H13 |
108.801 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability