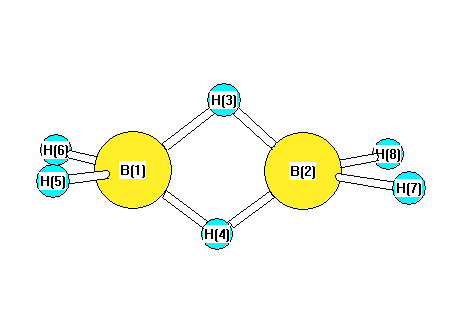
Vibrational Frequencies calculated at CCD/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2699 |
2544 |
0.00 |
|
|
|
| 2 |
Ag |
2261 |
2131 |
0.00 |
|
|
|
| 3 |
Ag |
1249 |
1177 |
0.00 |
|
|
|
| 4 |
Ag |
840 |
791 |
0.00 |
|
|
|
| 5 |
Au |
866 |
816 |
0.00 |
|
|
|
| 6 |
B1g |
2778 |
2618 |
0.00 |
|
|
|
| 7 |
B1g |
966 |
911 |
0.00 |
|
|
|
| 8 |
B1u |
2079 |
1959 |
19.33 |
|
|
|
| 9 |
B1u |
1013 |
955 |
22.25 |
|
|
|
| 10 |
B2g |
1947 |
1834 |
0.00 |
|
|
|
| 11 |
B2g |
882 |
831 |
0.00 |
|
|
|
| 12 |
B2u |
2792 |
2631 |
204.46 |
|
|
|
| 13 |
B2u |
1022 |
963 |
5.63 |
|
|
|
| 14 |
B2u |
387 |
364 |
12.50 |
|
|
|
| 15 |
B3g |
1083 |
1021 |
0.00 |
|
|
|
| 16 |
B3u |
2683 |
2528 |
161.23 |
|
|
|
| 17 |
B3u |
1815 |
1711 |
553.01 |
|
|
|
| 18 |
B3u |
1238 |
1167 |
90.67 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14300.1 cm
-1
Scaled (by 0.9423) Zero Point Vibrational Energy (zpe) 13475.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.