Jump to
S1C2
Vibrational Frequencies calculated at CCD/cc-pVDZ
Geometric Data calculated at CCD/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCD/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -231.806800 |
| Energy at 298.15K | -231.815120 |
| Nuclear repulsion energy | 175.286878 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCD/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3195 |
3057 |
9.37 |
|
|
|
| 2 |
A |
3174 |
3037 |
21.97 |
|
|
|
| 3 |
A |
3168 |
3031 |
21.28 |
|
|
|
| 4 |
A |
3152 |
3016 |
11.21 |
|
|
|
| 5 |
A |
3101 |
2967 |
10.70 |
|
|
|
| 6 |
A |
3082 |
2949 |
18.34 |
|
|
|
| 7 |
A |
3073 |
2940 |
12.24 |
|
|
|
| 8 |
A |
3060 |
2927 |
15.99 |
|
|
|
| 9 |
A |
1868 |
1787 |
112.79 |
|
|
|
| 10 |
A |
1501 |
1436 |
8.59 |
|
|
|
| 11 |
A |
1495 |
1431 |
5.83 |
|
|
|
| 12 |
A |
1482 |
1418 |
8.67 |
|
|
|
| 13 |
A |
1472 |
1408 |
15.00 |
|
|
|
| 14 |
A |
1461 |
1398 |
0.24 |
|
|
|
| 15 |
A |
1431 |
1369 |
14.58 |
|
|
|
| 16 |
A |
1414 |
1353 |
37.45 |
|
|
|
| 17 |
A |
1385 |
1325 |
4.65 |
|
|
|
| 18 |
A |
1291 |
1235 |
0.13 |
|
|
|
| 19 |
A |
1211 |
1159 |
59.71 |
|
|
|
| 20 |
A |
1140 |
1091 |
0.06 |
|
|
|
| 21 |
A |
1124 |
1075 |
0.52 |
|
|
|
| 22 |
A |
1020 |
976 |
3.53 |
|
|
|
| 23 |
A |
961 |
920 |
5.48 |
|
|
|
| 24 |
A |
956 |
915 |
2.41 |
|
|
|
| 25 |
A |
787 |
753 |
1.83 |
|
|
|
| 26 |
A |
763 |
730 |
2.02 |
|
|
|
| 27 |
A |
598 |
572 |
11.85 |
|
|
|
| 28 |
A |
480 |
459 |
0.12 |
|
|
|
| 29 |
A |
404 |
386 |
4.00 |
|
|
|
| 30 |
A |
253 |
242 |
4.34 |
|
|
|
| 31 |
A |
223 |
214 |
0.12 |
|
|
|
| 32 |
A |
120 |
114 |
0.02 |
|
|
|
| 33 |
A |
39 |
37 |
0.08 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24941.3 cm
-1
Scaled (by 0.9567) Zero Point Vibrational Energy (zpe) 23861.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
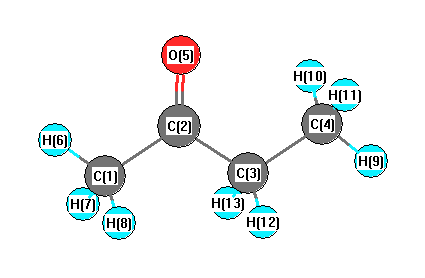
Geometric Data calculated at CCD/cc-pVDZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.896 |
-0.496 |
0.000 |
| C2 |
0.527 |
0.171 |
-0.000 |
| C3 |
-0.681 |
-0.765 |
-0.000 |
| C4 |
-2.017 |
-0.021 |
0.000 |
| O5 |
0.401 |
1.378 |
-0.000 |
| H6 |
2.685 |
0.272 |
-0.002 |
| H7 |
2.006 |
-1.140 |
0.892 |
| H8 |
2.005 |
-1.145 |
-0.888 |
| H9 |
-2.861 |
-0.733 |
-0.002 |
| H10 |
-2.104 |
0.625 |
0.890 |
| H11 |
-2.103 |
0.628 |
-0.887 |
| H12 |
-0.594 |
-1.431 |
-0.882 |
| H13 |
-0.593 |
-1.431 |
0.881 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5234 | 2.5914 | 3.9426 | 2.3975 | 1.1006 | 1.1051 | 1.1051 | 4.7633 | 4.2492 | 4.2481 | 2.8021 | 2.8014 |
C2 | 1.5234 | | 1.5279 | 2.5516 | 1.2135 | 2.1602 | 2.1683 | 2.1688 | 3.5063 | 2.8147 | 2.8128 | 2.1444 | 2.1441 | C3 | 2.5914 | 1.5279 | | 1.5296 | 2.4005 | 3.5218 | 2.8560 | 2.8542 | 2.1803 | 2.1796 | 2.1795 | 1.1082 | 1.1082 | C4 | 3.9426 | 2.5516 | 1.5296 | | 2.7939 | 4.7113 | 4.2704 | 4.2699 | 1.1036 | 1.1030 | 1.1030 | 2.1893 | 2.1893 | O5 | 2.3975 | 1.2135 | 2.4005 | 2.7939 | | 2.5374 | 3.1161 | 3.1185 | 3.8853 | 2.7635 | 2.7602 | 3.1075 | 3.1072 | H6 | 1.1006 | 2.1602 | 3.5218 | 4.7113 | 2.5374 | | 1.8035 | 1.8037 | 5.6360 | 4.8844 | 4.8818 | 3.7975 | 3.7980 | H7 | 1.1051 | 2.1683 | 2.8560 | 4.2704 | 3.1161 | 1.8035 | | 1.7797 | 4.9653 | 4.4733 | 4.8141 | 3.1605 | 2.6156 | H8 | 1.1051 | 2.1688 | 2.8542 | 4.2699 | 3.1185 | 1.8037 | 1.7797 | | 4.9631 | 4.8146 | 4.4742 | 2.6143 | 3.1561 | H9 | 4.7633 | 3.5063 | 2.1803 | 1.1036 | 3.8853 | 5.6360 | 4.9653 | 4.9631 | | 1.7920 | 1.7919 | 2.5304 | 2.5320 | H10 | 4.2492 | 2.8147 | 2.1796 | 1.1030 | 2.7635 | 4.8844 | 4.4733 | 4.8146 | 1.7920 | | 1.7775 | 3.1062 | 2.5517 | H11 | 4.2481 | 2.8128 | 2.1795 | 1.1030 | 2.7602 | 4.8818 | 4.8141 | 4.4742 | 1.7919 | 1.7775 | | 2.5533 | 3.1061 | H12 | 2.8021 | 2.1444 | 1.1082 | 2.1893 | 3.1075 | 3.7975 | 3.1605 | 2.6143 | 2.5304 | 3.1062 | 2.5533 | | 1.7626 | H13 | 2.8014 | 2.1441 | 1.1082 | 2.1893 | 3.1072 | 3.7980 | 2.6156 | 3.1561 | 2.5320 | 2.5517 | 3.1061 | 1.7626 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
116.263 |
|
C1 |
C2 |
O5 |
121.919 |
| C2 |
C1 |
H6 |
109.779 |
|
C2 |
C1 |
H7 |
110.149 |
| C2 |
C1 |
H8 |
110.191 |
|
C2 |
C3 |
C4 |
113.128 |
| C2 |
C3 |
H12 |
107.810 |
|
C2 |
C3 |
H13 |
107.788 |
| C3 |
C2 |
O5 |
121.818 |
|
C3 |
C4 |
H9 |
110.748 |
| C3 |
C4 |
H10 |
110.728 |
|
C3 |
C4 |
H11 |
110.717 |
| C4 |
C3 |
H12 |
111.191 |
|
C4 |
C3 |
H13 |
111.184 |
| H6 |
C1 |
H7 |
109.697 |
|
H6 |
C1 |
H8 |
109.725 |
| H7 |
C1 |
H8 |
107.263 |
|
H9 |
C4 |
H10 |
108.598 |
| H9 |
C4 |
H11 |
108.587 |
|
H10 |
C4 |
H11 |
107.359 |
| H12 |
C3 |
H13 |
105.360 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability