Jump to
S1C2
Vibrational Frequencies calculated at CCD/cc-pVTZ
Geometric Data calculated at CCD/cc-pVTZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCD/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -997.903296 |
| Energy at 298.15K | -997.908186 |
| HF Energy | -997.132395 |
| Nuclear repulsion energy | 202.638066 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCD/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3175 |
2964 |
1.10 |
|
|
|
| 2 |
A |
3121 |
2914 |
22.53 |
|
|
|
| 3 |
A |
1499 |
1399 |
0.29 |
|
|
|
| 4 |
A |
1370 |
1280 |
16.53 |
|
|
|
| 5 |
A |
1253 |
1170 |
0.58 |
|
|
|
| 6 |
A |
1077 |
1006 |
1.23 |
|
|
|
| 7 |
A |
980 |
915 |
9.39 |
|
|
|
| 8 |
A |
696 |
650 |
15.24 |
|
|
|
| 9 |
A |
265 |
247 |
0.75 |
|
|
|
| 10 |
A |
118 |
110 |
0.86 |
|
|
|
| 11 |
B |
3187 |
2976 |
4.65 |
|
|
|
| 12 |
B |
3113 |
2906 |
3.40 |
|
|
|
| 13 |
B |
1496 |
1397 |
10.11 |
|
|
|
| 14 |
B |
1347 |
1258 |
35.00 |
|
|
|
| 15 |
B |
1187 |
1108 |
0.68 |
|
|
|
| 16 |
B |
920 |
859 |
14.65 |
|
|
|
| 17 |
B |
723 |
675 |
19.85 |
|
|
|
| 18 |
B |
417 |
389 |
6.71 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 12971.1 cm
-1
Scaled (by 0.9337) Zero Point Vibrational Energy (zpe) 12111.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCD/cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.297 |
0.695 |
0.892 |
| C2 |
-0.297 |
-0.695 |
0.892 |
| Cl3 |
-0.297 |
1.685 |
-0.469 |
| Cl4 |
0.297 |
-1.685 |
-0.469 |
| H5 |
0.011 |
1.208 |
1.807 |
| H6 |
1.379 |
0.658 |
0.821 |
| H7 |
-0.011 |
-1.208 |
1.807 |
| H8 |
-1.379 |
-0.658 |
0.821 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
Cl3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.5115 | 1.7844 | 2.7412 | 1.0873 | 1.0854 | 2.1340 | 2.1553 |
C2 | 1.5115 | | 2.7412 | 1.7844 | 2.1340 | 2.1553 | 1.0873 | 1.0854 | Cl3 | 1.7844 | 2.7412 | | 3.4210 | 2.3455 | 2.3513 | 3.6918 | 2.8848 | Cl4 | 2.7412 | 1.7844 | 3.4210 | | 3.6918 | 2.8848 | 2.3455 | 2.3513 | H5 | 1.0873 | 2.1340 | 2.3455 | 3.6918 | | 1.7740 | 2.4165 | 2.5274 | H6 | 1.0854 | 2.1553 | 2.3513 | 2.8848 | 1.7740 | | 2.5274 | 3.0565 | H7 | 2.1340 | 1.0873 | 3.6918 | 2.3455 | 2.4165 | 2.5274 | | 1.7740 | H8 | 2.1553 | 1.0854 | 2.8848 | 2.3513 | 2.5274 | 3.0565 | 1.7740 | |
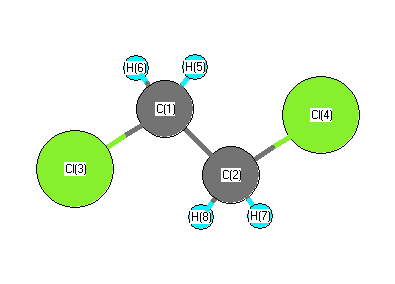 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl4 |
112.279 |
|
C1 |
C2 |
H7 |
109.312 |
| C1 |
C2 |
H8 |
111.124 |
|
C2 |
C1 |
Cl3 |
112.279 |
| C2 |
C1 |
H5 |
109.312 |
|
C2 |
C1 |
H6 |
111.124 |
| Cl3 |
C1 |
H5 |
107.008 |
|
Cl3 |
C1 |
H6 |
107.525 |
| Cl4 |
C2 |
H7 |
107.008 |
|
Cl4 |
C2 |
H8 |
107.525 |
| H5 |
C1 |
H6 |
109.474 |
|
H7 |
C2 |
H8 |
109.474 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability