Jump to
S1C2
Energy calculated at CCD/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -323.076302 |
| Energy at 298.15K | |
| HF Energy | -321.862106 |
| Nuclear repulsion energy | 235.943850 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCD/cc-pVTZ
Geometric Data calculated at CCD/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.910 |
2.191 |
0.000 |
| C2 |
-1.506 |
0.719 |
0.000 |
| C3 |
0.000 |
0.566 |
0.000 |
| O4 |
0.294 |
-0.835 |
0.000 |
| N5 |
1.665 |
-1.018 |
0.000 |
| O6 |
1.935 |
-2.156 |
0.000 |
| H7 |
-2.993 |
2.298 |
0.000 |
| H8 |
-1.524 |
2.705 |
0.881 |
| H9 |
-1.524 |
2.705 |
-0.881 |
| H10 |
-1.913 |
0.214 |
-0.877 |
| H11 |
-1.913 |
0.214 |
0.877 |
| H12 |
0.437 |
1.030 |
0.886 |
| H13 |
0.437 |
1.030 |
-0.886 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5258 | 2.5069 | 3.7432 | 4.8029 | 5.8031 | 1.0886 | 1.0907 | 1.0907 | 2.1625 | 2.1625 | 2.7641 | 2.7641 |
C2 | 1.5258 | | 1.5141 | 2.3786 | 3.6155 | 4.4846 | 2.1685 | 2.1724 | 2.1724 | 1.0905 | 1.0905 | 2.1585 | 2.1585 | C3 | 2.5069 | 1.5141 | | 1.4322 | 2.2979 | 3.3403 | 3.4576 | 2.7696 | 2.7696 | 2.1337 | 2.1337 | 1.0910 | 1.0910 | O4 | 3.7432 | 2.3786 | 1.4322 | | 1.3827 | 2.1067 | 4.5410 | 4.0758 | 4.0758 | 2.5962 | 2.5962 | 2.0696 | 2.0696 | N5 | 4.8029 | 3.6155 | 2.2979 | 1.3827 | | 1.1701 | 5.7171 | 4.9798 | 4.9798 | 3.8839 | 3.8839 | 2.5460 | 2.5460 | O6 | 5.8031 | 4.4846 | 3.3403 | 2.1067 | 1.1701 | | 6.6426 | 6.0306 | 6.0306 | 4.6038 | 4.6038 | 3.6300 | 3.6300 | H7 | 1.0886 | 2.1685 | 3.4576 | 4.5410 | 5.7171 | 6.6426 | | 1.7607 | 1.7607 | 2.5056 | 2.5056 | 3.7629 | 3.7629 | H8 | 1.0907 | 2.1724 | 2.7696 | 4.0758 | 4.9798 | 6.0306 | 1.7607 | | 1.7626 | 3.0734 | 2.5210 | 2.5791 | 3.1263 | H9 | 1.0907 | 2.1724 | 2.7696 | 4.0758 | 4.9798 | 6.0306 | 1.7607 | 1.7626 | | 2.5210 | 3.0734 | 3.1263 | 2.5791 | H10 | 2.1625 | 1.0905 | 2.1337 | 2.5962 | 3.8839 | 4.6038 | 2.5056 | 3.0734 | 2.5210 | | 1.7533 | 3.0489 | 2.4881 | H11 | 2.1625 | 1.0905 | 2.1337 | 2.5962 | 3.8839 | 4.6038 | 2.5056 | 2.5210 | 3.0734 | 1.7533 | | 2.4881 | 3.0489 | H12 | 2.7641 | 2.1585 | 1.0910 | 2.0696 | 2.5460 | 3.6300 | 3.7629 | 2.5791 | 3.1263 | 3.0489 | 2.4881 | | 1.7712 | H13 | 2.7641 | 2.1585 | 1.0910 | 2.0696 | 2.5460 | 3.6300 | 3.7629 | 3.1263 | 2.5791 | 2.4881 | 3.0489 | 1.7712 | |
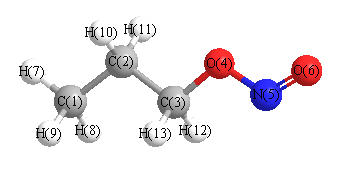 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.107 |
|
C1 |
C2 |
H10 |
110.386 |
| C1 |
C2 |
H11 |
110.386 |
|
C2 |
C1 |
H7 |
110.976 |
| C2 |
C1 |
H8 |
111.164 |
|
C2 |
C1 |
H9 |
111.164 |
| C2 |
C3 |
O4 |
107.632 |
|
C2 |
C3 |
H12 |
110.859 |
| C2 |
C3 |
H13 |
110.859 |
|
C3 |
C2 |
H10 |
108.923 |
| C3 |
C2 |
H11 |
108.923 |
|
C3 |
O4 |
N5 |
109.424 |
| O4 |
C3 |
H12 |
109.471 |
|
O4 |
C3 |
H13 |
109.471 |
| O4 |
N5 |
O6 |
110.958 |
|
H7 |
C1 |
H8 |
107.786 |
| H7 |
C1 |
H9 |
107.786 |
|
H8 |
C1 |
H9 |
107.806 |
| H10 |
C2 |
H11 |
107.008 |
|
H12 |
C3 |
H13 |
108.532 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCD/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -323.074423 |
| Energy at 298.15K | |
| HF Energy | -321.856960 |
| Nuclear repulsion energy | 238.590611 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCD/cc-pVTZ
Geometric Data calculated at CCD/cc-pVTZ
Point Group is C1
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability