Jump to
S1C2
Energy calculated at wB97X-D/6-31G*
| | hartrees |
|---|
| Energy at 0K | -897.091437 |
| Energy at 298.15K | -897.094593 |
| HF Energy | -897.091437 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
908 |
861 |
142.56 |
|
|
|
| 2 |
A1 |
646 |
613 |
6.24 |
|
|
|
| 3 |
A1 |
513 |
486 |
32.73 |
|
|
|
| 4 |
B1 |
430 |
408 |
0.00 |
|
|
|
| 5 |
B2 |
611 |
579 |
0.00 |
|
|
|
| 6 |
B2 |
216 |
205 |
0.00 |
|
|
|
| 7 |
E |
862 |
817 |
420.33 |
|
|
|
| 7 |
E |
862 |
817 |
420.33 |
|
|
|
| 8 |
E |
487 |
462 |
3.56 |
|
|
|
| 8 |
E |
487 |
462 |
3.56 |
|
|
|
| 9 |
E |
332 |
315 |
0.10 |
|
|
|
| 9 |
E |
332 |
315 |
0.10 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3341.5 cm
-1
Scaled (by 0.9485) Zero Point Vibrational Energy (zpe) 3169.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-31G*
Point Group is C4v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.000 |
0.213 |
| F2 |
0.000 |
0.000 |
-1.360 |
| F3 |
0.000 |
1.626 |
0.245 |
| F4 |
-1.626 |
0.000 |
0.245 |
| F5 |
0.000 |
-1.626 |
0.245 |
| F6 |
1.626 |
0.000 |
0.245 |
Atom - Atom Distances (Å)
| |
S1 |
F2 |
F3 |
F4 |
F5 |
F6 |
| S1 | | 1.5730 | 1.6261 | 1.6261 | 1.6261 | 1.6261 |
F2 | 1.5730 | | 2.2850 | 2.2850 | 2.2850 | 2.2850 | F3 | 1.6261 | 2.2850 | | 2.2991 | 3.2514 | 2.2991 | F4 | 1.6261 | 2.2850 | 2.2991 | | 2.2991 | 3.2514 | F5 | 1.6261 | 2.2850 | 3.2514 | 2.2991 | | 2.2991 | F6 | 1.6261 | 2.2850 | 2.2991 | 3.2514 | 2.2991 | |
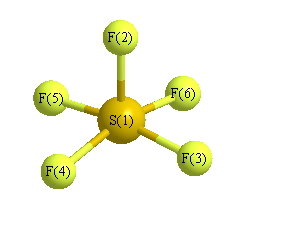 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| F2 |
S1 |
F3 |
91.151 |
|
F2 |
S1 |
F4 |
91.151 |
| F2 |
S1 |
F5 |
91.151 |
|
F2 |
S1 |
F6 |
91.151 |
| F3 |
S1 |
F4 |
89.977 |
|
F3 |
S1 |
F5 |
177.699 |
| F3 |
S1 |
F6 |
89.977 |
|
F4 |
S1 |
F5 |
89.977 |
| F4 |
S1 |
F6 |
177.699 |
|
F5 |
S1 |
F6 |
89.977 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
1.535 |
|
|
|
| 2 |
F |
-0.275 |
|
|
|
| 3 |
F |
-0.315 |
|
|
|
| 4 |
F |
-0.315 |
|
|
|
| 5 |
F |
-0.315 |
|
|
|
| 6 |
F |
-0.315 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.450 |
0.450 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.314 |
0.000 |
0.000 |
| y |
0.000 |
-38.314 |
0.000 |
| z |
0.000 |
0.000 |
-34.910 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.702 |
0.000 |
0.000 |
| y |
0.000 |
-1.702 |
0.000 |
| z |
0.000 |
0.000 |
3.404 |
|
| Polar |
|---|
| 3z2-r2 | 6.809 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.929 |
0.000 |
0.000 |
| y |
0.000 |
3.929 |
0.000 |
| z |
0.000 |
0.000 |
2.779 |
<r2> (average value of r
2) Å
2
| <r2> |
138.232 |
| (<r2>)1/2 |
11.757 |
Jump to
S1C1
Energy calculated at wB97X-D/6-31G*
| | hartrees |
|---|
| Energy at 0K | -897.091211 |
| Energy at 298.15K | -897.094385 |
| HF Energy | -897.091211 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
903 |
857 |
141.75 |
|
|
|
| 2 |
A' |
861 |
817 |
414.64 |
|
|
|
| 3 |
A' |
646 |
613 |
7.34 |
|
|
|
| 4 |
A' |
612 |
580 |
0.03 |
|
|
|
| 5 |
A' |
517 |
491 |
32.19 |
|
|
|
| 6 |
A' |
484 |
459 |
3.52 |
|
|
|
| 7 |
A' |
326 |
309 |
0.08 |
|
|
|
| 8 |
A' |
219 |
208 |
0.01 |
|
|
|
| 9 |
A" |
865 |
821 |
416.45 |
|
|
|
| 10 |
A" |
499 |
473 |
4.48 |
|
|
|
| 11 |
A" |
431 |
409 |
0.00 |
|
|
|
| 12 |
A" |
339 |
322 |
0.18 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3351.0 cm
-1
Scaled (by 0.9485) Zero Point Vibrational Energy (zpe) 3178.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.196 |
0.083 |
0.000 |
| F2 |
-1.384 |
0.033 |
0.000 |
| F3 |
0.401 |
1.595 |
0.000 |
| F4 |
0.212 |
0.118 |
1.670 |
| F5 |
0.212 |
-2.012 |
0.000 |
| F6 |
0.212 |
0.118 |
-1.670 |
Atom - Atom Distances (Å)
| |
S1 |
F2 |
F3 |
F4 |
F5 |
F6 |
| S1 | | 1.5809 | 1.5258 | 1.6699 | 2.0951 | 1.6699 |
F2 | 1.5809 | | 2.3723 | 2.3110 | 2.5933 | 2.3110 | F3 | 1.5258 | 2.3723 | | 2.2375 | 3.6120 | 2.2375 | F4 | 1.6699 | 2.3110 | 2.2375 | | 2.7059 | 3.3390 | F5 | 2.0951 | 2.5933 | 3.6120 | 2.7059 | | 2.7059 | F6 | 1.6699 | 2.3110 | 2.2375 | 3.3390 | 2.7059 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| F2 |
S1 |
F3 |
99.550 |
|
F2 |
S1 |
F4 |
90.575 |
| F2 |
S1 |
F5 |
88.585 |
|
F2 |
S1 |
F6 |
90.575 |
| F3 |
S1 |
F4 |
88.759 |
|
F3 |
S1 |
F5 |
171.865 |
| F3 |
S1 |
F6 |
88.759 |
|
F4 |
S1 |
F5 |
91.175 |
| F4 |
S1 |
F6 |
177.408 |
|
F5 |
S1 |
F6 |
91.175 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
1.535 |
|
|
|
| 2 |
F |
-0.278 |
|
|
|
| 3 |
F |
-0.314 |
|
|
|
| 4 |
F |
-0.316 |
|
|
|
| 5 |
F |
-0.312 |
|
|
|
| 6 |
F |
-0.316 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.525 |
0.056 |
0.000 |
0.528 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.926 |
-0.111 |
0.000 |
| y |
-0.111 |
-38.239 |
0.000 |
| z |
0.000 |
0.000 |
-38.327 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.357 |
-0.111 |
0.000 |
| y |
-0.111 |
-1.612 |
0.000 |
| z |
0.000 |
0.000 |
-1.745 |
|
| Polar |
|---|
| 3z2-r2 | -3.489 |
|---|
| x2-y2 | 3.313 |
|---|
| xy | -0.111 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.779 |
0.035 |
0.000 |
| y |
0.035 |
3.939 |
0.000 |
| z |
0.000 |
0.000 |
3.918 |
<r2> (average value of r
2) Å
2
| <r2> |
138.031 |
| (<r2>)1/2 |
11.749 |