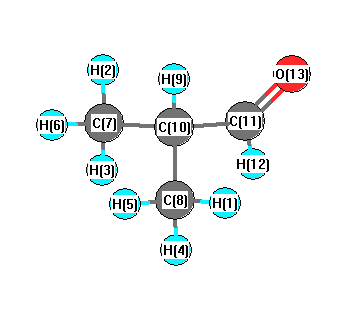
Vibrational Frequencies calculated at wB97X-D/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3136 |
3000 |
26.73 |
|
|
|
| 2 |
A' |
3124 |
2989 |
48.08 |
|
|
|
| 3 |
A' |
3088 |
2954 |
3.59 |
|
|
|
| 4 |
A' |
3049 |
2917 |
18.39 |
|
|
|
| 5 |
A' |
2863 |
2738 |
98.81 |
|
|
|
| 6 |
A' |
1849 |
1769 |
233.36 |
|
|
|
| 7 |
A' |
1517 |
1451 |
17.36 |
|
|
|
| 8 |
A' |
1514 |
1448 |
8.25 |
|
|
|
| 9 |
A' |
1438 |
1375 |
0.57 |
|
|
|
| 10 |
A' |
1422 |
1360 |
4.28 |
|
|
|
| 11 |
A' |
1328 |
1270 |
0.73 |
|
|
|
| 12 |
A' |
1193 |
1141 |
9.22 |
|
|
|
| 13 |
A' |
1189 |
1138 |
0.12 |
|
|
|
| 14 |
A' |
931 |
891 |
19.52 |
|
|
|
| 15 |
A' |
856 |
819 |
19.44 |
|
|
|
| 16 |
A' |
553 |
529 |
5.50 |
|
|
|
| 17 |
A' |
361 |
345 |
7.54 |
|
|
|
| 18 |
A' |
339 |
324 |
0.45 |
|
|
|
| 19 |
A' |
244 |
233 |
0.76 |
|
|
|
| 20 |
A" |
3135 |
2999 |
11.22 |
|
|
|
| 21 |
A" |
3118 |
2983 |
7.11 |
|
|
|
| 22 |
A" |
3048 |
2915 |
22.17 |
|
|
|
| 23 |
A" |
1501 |
1436 |
2.61 |
|
|
|
| 24 |
A" |
1491 |
1427 |
0.26 |
|
|
|
| 25 |
A" |
1406 |
1345 |
5.00 |
|
|
|
| 26 |
A" |
1345 |
1286 |
0.57 |
|
|
|
| 27 |
A" |
1146 |
1096 |
1.34 |
|
|
|
| 28 |
A" |
994 |
951 |
0.33 |
|
|
|
| 29 |
A" |
960 |
918 |
0.23 |
|
|
|
| 30 |
A" |
935 |
894 |
1.56 |
|
|
|
| 31 |
A" |
331 |
317 |
0.89 |
|
|
|
| 32 |
A" |
219 |
209 |
0.03 |
|
|
|
| 33 |
A" |
30 |
28 |
7.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24825.1 cm
-1
Scaled (by 0.9566) Zero Point Vibrational Energy (zpe) 23747.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.258 |
|
|
|
| 2 |
H |
0.258 |
|
|
|
| 3 |
H |
0.276 |
|
|
|
| 4 |
H |
0.276 |
|
|
|
| 5 |
H |
0.253 |
|
|
|
| 6 |
H |
0.253 |
|
|
|
| 7 |
C |
-1.038 |
|
|
|
| 8 |
C |
-1.038 |
|
|
|
| 9 |
H |
0.222 |
|
|
|
| 10 |
C |
0.640 |
|
|
|
| 11 |
C |
0.108 |
|
|
|
| 12 |
H |
0.351 |
|
|
|
| 13 |
O |
-0.818 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.488 |
2.956 |
0.000 |
2.996 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.763 |
1.183 |
0.000 |
| y |
1.183 |
-38.681 |
0.000 |
| z |
0.000 |
0.000 |
-30.848 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.002 |
1.183 |
0.000 |
| y |
1.183 |
-7.876 |
0.000 |
| z |
0.000 |
0.000 |
3.874 |
|
| Polar |
|---|
| 3z2-r2 | 7.748 |
|---|
| x2-y2 | 7.919 |
|---|
| xy | 1.183 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.446 |
-0.145 |
0.000 |
| y |
-0.145 |
8.937 |
0.000 |
| z |
0.000 |
0.000 |
7.606 |
<r2> (average value of r
2) Å
2
| <r2> |
134.011 |
| (<r2>)1/2 |
11.576 |